鉑-鈷雙核配合物:合成、光物理性質(zhì)及可見光驅(qū)動分解水制氫
2013-09-15 03:03:40韓阿麗杜平武
無機化學學報 2013年8期
韓阿麗 杜平武
(中國科學技術(shù)大學化學與材料科學學院,合肥 230026)
0 Introduction
Visible light-d riven water splitting is a great challenge and an attractive topic for solar energy conversion.The reduction side of this reaction to generate hydrogen fuel has been studied since 1970s and many proof-of-concept systems for hydrogen production have been reported[1-2].Such systems generally contain a sacrificial electron donor,a photosensitizer (such as [Ru (bpy)3]2+,bpy=2,2′-bipyridine),an electron relay and a metal colloidal catalyst.Recently,the discovery of long-lived excited states of platinum terpyridyl acetylide photosensitizers and iridium cyclometalated photosensitizers has led to the development of more efficient multi-component systems to replace[Ru(bpy)3]2+for hydrogen production with both higher turnover frequencies(TOF)and overall turnover numbers (TONs)[3-5].However,these systems also rely preferably on expensive and unsustainable noble metals as the catalysts.Recently,many different catalysts based on earth-abundant elements such as Ni,Co and Fe have been reported for hydrogen production in electrocatalytic or photocatalytic systems[6-7].Cobaloxime(scheme 1)represents one example of Co based molecular catalysts and attracts much attention for visible light-driven hydrogen production from water in the presence of a photosensitizer and a sacrificial electron donor,in which the reaction mechanism has alsobeen discussed[8-14].
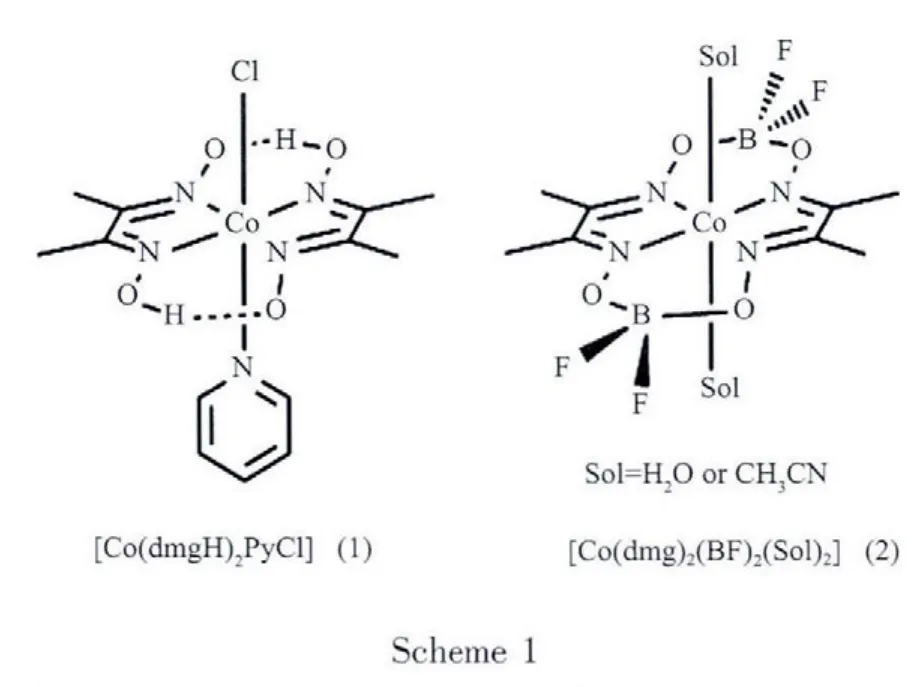
In contrast with the multi-component systems,many molecular photochemical devices by covalently or coordinately linking a photosensitizer with a molecular catalyst have been constructed with the aim of higher efficiency for hydrogen production because electron transfer processes would be possibly more favorable in the devices.Various photosensitizer-catalyst dyads have been reported as molecular photochemical devices for hydrogen production.In some of the systems reported,a Pt(Ⅱ) or Pd(Ⅱ) molecular catalyst is covalently linked to therutheniumsensitizer without any electron relays[15-16].In the presence of a sacrificial donor,the systems do producehydrogen upon visible light irradiation and give TONs as 4.8 and 56 for Pt(Ⅱ) and Pd(Ⅱ),respectively.Furthermore,a Rh(Ⅱ)catalyst was directly attached to the ruthenium photosensitizer to make a molecular device,[{(bpy)2Ru(dpp)}2RhCl2](PF6)5(bpy=bipyridine,dpp =2,3-bis (2-pyridyl)pyrazine), for hydrogen production[17].Artero and co-workers[18-19]employed[Ru(bpy)3]2+with cobaloximes (1 and 2)or iridiumcobaloxime system for photochemical H2generation in organic solvents,but system efficiency decreased dramatically in the presence of small amounts of water.Sun and co-workers[8]examined a Znic porphyrin-[2Fe-2S]system for a TONs of 22 under visible light.Some other photosensitizer-catalyst hybrids could also be found in recent literatures[14,21].
In this contribution,we report the photophysical properties and visible light-driven hydrogen production from water based on a heterodinuclear platinum-cobalt complex.
1 Experimental
1.1 Materials
Acetonitrile, methanol, and triethanolamine(TEOA)were purchased (Sigma-Aldrich)and used without further purification. [Pt(tBu3tpy)Cl]PF6was synthesized following the reported procedure[22].All of the new complexes were synthesized,and were characterized by1H-NMR,ESI-MS spectrometry and elemental analysis.
1.2 Synthesis of complex 3 and complex 5
[Pt(tBu3tpy)([C≡C-C6H4N])](3,tBu3tpy=4,4′,4′′-tri(tert-butyl)-2,2′,6′,2′′-terpyridine).5 mg CuI,5 mL triethylamine(TEA)and 10 mg 4-Ethynylpyridine were added in 10 mL dimethylformamide(DMF),followed by addition of 50 mg[Pt(tBu3tpy)Cl]PF6(0.064 mmol).The mixture was stirred at room temperature for 3 d.TEA was then evaporated and the DMF solution was filtered into an aqueous solution of NH4PF6.The orange precipitate was collected by a frit and washed twice by methanol and diethyl ether to yield a reddish orange solid(38 mg,yield,71%).1H NMR (DMSO-d6,400.1 MHz):8.97(2H,d),8.72(4H,m),7.89(d,2H),7.75(d,2H),7.46(d,2H),1.50(s,9H),1.40(s,18H)ESI-MS Calcd.for M+(C34H39N4Pt)698.79,Found 698.67(M+).
Complex 5.Complex 3(30 mg,0.036 mmol)and complex 4(13 mg,0.036 mmol)were added in 10 mL methanol followed by addition of 1 equivalent triethylamine.The mixture was stirred at room temperature for 6 h and the yellow precipitate was collected by a frit.The crude product was washed by methanol and diethyl ether twice to yield a product of 29 mg(yield:80%).1H NMR(DMSO-d6,400.1 MHz):8.82(2H,d),8.70(4H,m),7.95(d,2H),7.80(d,2H),7.43(d,2H),2.35(s,12H),1.50(s,9H),1.40(s,18H).Infrared Spectra(IR,cm-1):3 438.52(broad),2 965.54,2 118.51,1 610.96,1 563.19,1 474.66,1 425.30,1 372.21,1 241.29,1 092.96,1 028.87,840.88.ESIMS Calcd.for M+(C42H53ClCoN8O4Pt)1023.40,Found:1022.83.Anal.Calcd.(C42H53ClCoN8O4PtPF6·MeOH):C,43.02;H,4.79;N,9.33.Found:C,42.53;H,4.35;N,9.17.
1.3 Photophysical propertiesand photocatalysis
Absorption spectra were recorded using a Hitachi U2000 scanning spectrophotometer(200~1 100 nm).Emission spectra were obtained using a Spex Fluoromax-P fluorometer corrected for instrument response.Monochromatorswere positioned with a 2-nm band-pass,and solution samples were degassed by at least three freeze-pump-thaw cycles before mesurements.
For photoinduced hydrogen evolution,each sample was made in a 100 mL round bottom flask with volume of 25 mL in acetonitrile:water(24 ∶1,V/V).Typically,the sample contained 4.0×10-5mol·L-1Pt chromophore or Pt-Co molecular catalyst.Hydrochloric acid or sodium hydroxide solution were used to adjust the pH value.The flask was sealed with a rubber septum and degassed by bubbling nitrogen for 15 min under atmospheric pressure at room temperature,after which 5 mL nitrogen was removed fromthe flask and replaced with 5 mL of methane (1.013 kpa)to serve as the internal standard.The samples were irradiated under a 200WMercury Xexon lamp.A cut-off filter was used to remove off light with λ<400 nm.The amount of H2generated was determined by GC analysis using a chromatograph (Shimadzu)with a molecular sieve 0.5 nm (30 m×0.53 mm,PLOT column,df=50 μm)and thermal conductivity detector.The gas samples were directly injected by a syringe(100μL/each)and non split stream was applied.The temperature for column is 60℃ and the temperature for detector is 90℃.The temperature for the injection port is90℃.Nitrogen was used asthe carrier gas and the rate is 30 mL·min-1.
2 Resultsand discussion
2.1 Photophysical propertiesof complex 3 and complex 5
The steady-state spectroscopy of complexes 3 and 5 were examined at room temperature.The spectroscopic data are consistent with a cationic Pt(Ⅱ)complex having a square-planar geometry with three of the four coordination sites occupied by the terpyridyl ligand and the fourth site occupied by acetylide[23-24].The UV-Vis absorption spectra of 3 and 5 measured in MeCN/H2O (24 ∶1,V/V)are shown in Fig.1(a).The absorption spectra exhibit a broad low energy absorption band between 371~480 nm with λmaxat 405 nm (ε~6800 dm3·mol-1·cm-1)for complex 3 and λmaxat 396 nm (ε~9300 dm3mol-1cm-1)for complex 5,which mainly corresponds to the dπ(Pt)-π(terpy)metal-toligand charge transfer (MLCT)transition based on previous assignments made for this class of complexes[23-24].The absorption bands in the range of 210~371 nmcorrespond to spin-allowed intraligand(ππ*)transitions(ε>2×104dm3·mol-1·cm-1).
The emission spectra was also investigated in MeCN/H2O (24:1,V/V)at room temperature.When complex 3 is excited at 400 nm (Fig.1 (b)),the emission spectra show a broad weak emission band from 480 nm to 700 nm with a maximum emission peak at 538 nm,giving a luminescence quantum yield (Φ)of~0.009,based on [Ru(bpy)3](PF6)2(Φ=0.062)[25]. According to previous photophysical studies of closely related [Pt(terpyridyl)(acetylide)]+complex[23-24],the excited state that gives rise to the observed emission in complex 3 is a triplet metal-toligand charge transfer (3MLCT)involving a dπ(Pt)metal orbital as the HOMO and theπ*(tBu3tpy)orbital as the LUMO.However,complex 5 is almost nonemissive (φ<0.000 5)in fluid solution,indicating the intramolecular electron transfer is very efficient from the Pt photosensitizer unit to the cobalt center.The emission property is consistent with previous observations made with the closely related dyad based on the Pt photosensitizers or Ru photosensitizers[26-27].

2.2 Visible light-driven hydrogen production
Using complex 5 as the hydrogen production catalyst,when TEOA is present as the sacrificial electron donor,the system does produce hydrogen upon visible light irradiation(long pass cut filter,λ>400 nm).The reaction condition is as follows:room temperature,4.0 ×10-5mol·L-1catalyst 5,1.60 ×10-2mol·L-1TEOA in MeCN/H2O(24∶1,V/V).Quantitative determination of generated H2was conducted by gas chromatography(GC)analysis with added methane as an internal standard.The turnover numbers(TONs)are defined with respect to the mole numbers of catalyst 5.
The production of hydrogen by photocatalyst 5 is impacted by many factors.At pH value of 8.5,hydrogen is generated with a turnover number of 105 after 10 hours(run 1).Within the same reaction time,the system gives a turnover number of 5 and 37 for pH value of 5.0 and 11(run 2 and 3),respectively.At higher pH values,the required redox potential for water reduction is increased,which might decrease the rate of hydrogen production.At lower pH values,the sacrificial electron donor of TEOA will be protonated and therefore weaken the electron donating ability,which could decrease the rate of hydrogen production from water.This observation is consistent with previous results for pH dependence of cobaloxime catalysts.[12]Control experiments indicate that all of the components-molecular catalyst 5,TEOA and water are essential for hydrogen production.The absence of any one of them yields insignificant amounts of hydrogen,as shown in Table 1,run 4~6.The activity of catalyst 5 also varies with different solvents (run 7~10),showing the following order for hydrogen production:MeCN>MeOH>acetone>DMF.

Table 1 Photocatalytic hydrogen production under different conditionsa
2.3 Mechanism of thephotocatalytic reaction
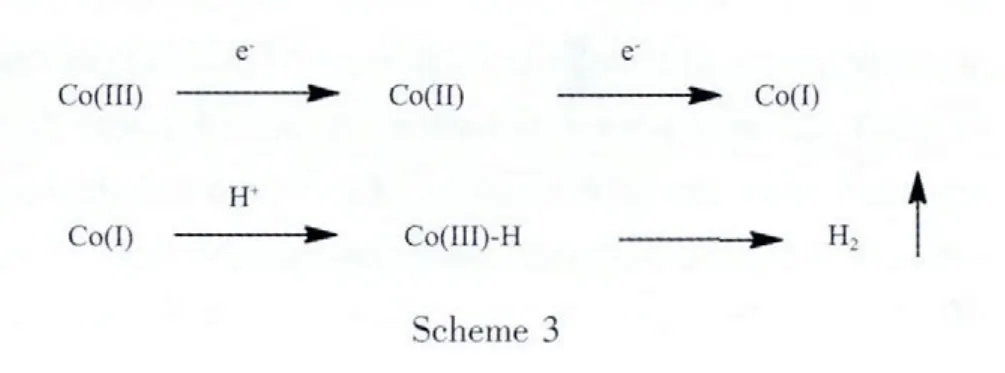
The mechanism of the visible light-driven hydrogen production in the multi-component system containing the Pt photosensitizer and the cobaloxime catalyst has been fully discussed in previous reports,in which the first step could be either oxidative quenching or reductivequenchingupon visiblelight irradiation[4c,13].In the present system,the emission spectra of the molecular catalyst 5 indicates the first step might be oxidative quenching via fast intramolecular electron transfer from the Pt photosensitizer part to the cobalt catalytic center[12b].Subsequently,the electron transfer from TEOA to the Pt photosensitizer unit may regenerate the ground state of the photosensitizer and prevent the non-productive back electron transfer.Further electron transfer from the Pt photosensitizer to Co(Ⅱ) catalytic center will produce the Coガ species,which are then protonated to give the important Coバhydride intermediates for further hydrogen production(Scheme 3).
Experimentally,the transformation of Co species could be monitored by UV-Vis spectroscopy in a solution containing complex 5 and TEOA at different pH values during visible light-driven catalytic reactions,as shown in Fig.2.At pH=8.5,the absorption spectra are shown in Fig.2(top).The spectra show a slight increase in the range between 420 nm and 520 nm,which istentatively assigned tothe absorption of Co(Ⅱ)species.At higher pH values,the system has a slower rate for hydrogen production but the Co(I)species could be observed in the range between 480 nm and 650 nm after 6 h irradiation,as shown in Fig.2(bottom)[28].
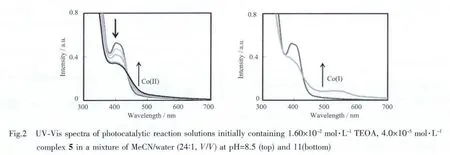
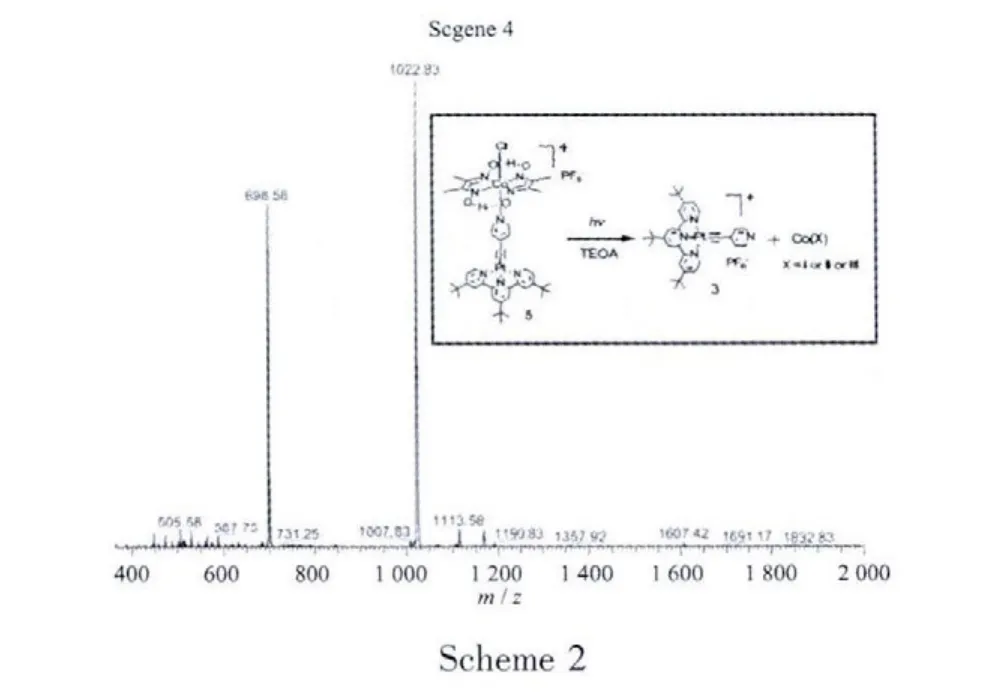
However,after carefully examining the absorption spectra before and after photoirradiation at pH=8.5,it seems that complex 5 might be decomposed during reaction.The final absorption spectrum in Fig.2(left)shows a decreasing extinction coefficient at maximum peak 405 nm,which is closer to complex 3 under the same concentration.It’s reasonable to propose that complex 3 is partly released from complex 5 during photoinduced electron transfer,as demonstrated in scheme 4 (inset).This process could be confirmed by mass spectrometry(scheme 4),which mainly shows two separatepeaksat 698.58(m/z of 3)and 1 022.83(m/z of 5)after 20 min photoirradiation.A recent paper has reported the unstability of a fluorecein-cobaloxime catalyst for photoinduced hydrogen production,[14]in which the first step is reductive quenching during photoirradiation.While oxidative quenching is the first step during photoirradiation,the present platinumcobalt catalyst shows another example for decompostion of photosensitizer-catalyst dyad during catalysis.
3 Conclusions
In conclusion,wehavedemonstrated photoinduced electron transfer process and visible light-driven H2production in a platinum-cobaloxime dyad.Mechanistic study shows the formation of Co(Ⅱ) and Coガ species upon irradiation.However,the dyad is probably decomposed to separate the photosensitizer and the catalyst.Further study is in progress to stabilize the cobalt catalytic center.
[1]Kalyanasundaram K,Kiwi J,Gratzel M.Helv.Chim.Acta,1978,61:2720-2730
[2]Esswein A J,Nocera D G.Chem.Rev.,2007,107:4022-4047
[3]Goldsmith JI,Hudson WR,Lowry M S,et al.J.Am.Chem.Soc.,2005,127:7502-7510
[4]Du P W,Schneider J,Jarosz P,et al.J.Am.Chem.Soc.,2006,128:7726-7727
[5]Du PW,Schneider J,Jarosz P,et al.J.Phys.Chem.B,2007,111:6887-6894
[6]Du P W,Eisenberg R.Energy Environ.Sci.,2012,5:6012-6021
[7]Artero V,Chavarot-Kerlidou M,Fontecave M.Angew.Chem.Int.Ed.,2011,50:7238-7266
[8]Razavet M,Artero V,Fontecave M.Inorg.Chem.,2005,44:4786-4795
[9]Baffert C,Artero V,Fontecave M.Inorg.Chem.,2007,46:1817-1824
[10]Du PW,Knowles K,Eisenberg R.J.Am.Chem.Soc.,2008,130:12576-12577
[11]Zhang P,Wang M,Dong J,et al.J.Phys.Chem.C,2010,114:15868-15874
[12]Hu X,Brunschwig B S,Peters JC.J.Am.Chem.Soc.,2007,129:8988-8998
[13]Hu X,Cossairt BM,Brunschwig BS,et al.Chem.Commun.,2005,37:4723-4725
[14]Dempsey J L,Brunschwig B S,Winkler J R,et al.Acc.Chem.Res.,2009,42:1995-2004
[15]Ozawa H,Haga M A,Sakai K.J.Am.Chem.Soc.,2006,128:4926-4927
[16]Rau S,Schfer B,Gleich D,et al.Angew.Chem.Int.Ed.,2006,45:6215-6218
[17]Elvington M,Brown J,Arachchige SM,et al.J.Am.Chem.Soc.,2007,129:10644-10645
[18]Fihri A,Artero V,Pereira A,et al.Dalton Trans.,2008,41:5567-5569
[19]Fihri A,Artero V,Razavet M,et al.Angew.Chem.Int.Ed.,2008,47:564-567
[20]Zhang P,Wang M,Li C,et al.Chem.Commun.,2010,46:8806-8808
[21]Artero V,Chavarot-Kerlidou M,Fontecave M.Angew.Chem.Int.Ed.,2011,50:7238-7266
[22]Lai SW,Chan M C W,Cheung K K,et al.Inorg.Chem.,1999,38:4262-4267
[23]Yam V W W,Tang R P L,Wong K M C,et al.Organometallics 2001,20:4476-4482.
[24]Yang Q Z,Wu L Z,Wu Z X,et al.Inorg.Chem.,2002,41:5653-5655.
[25]Calvert JM,Caspar JV,Binstead R A,et al.J.Am.Chem.Soc.,1982,104:6620-6627
[26]Chakraborty S,Wadas T J,Hester H,et al.Inorg.Chem.,2005,44:6284-6293
[27]Chakraborty S,Wadas T J,Hester H,et al.Inorg.Chem.,2005,44:6865-6878
[28]McCormick T M,Han Z,Weinberg D J,et al.Inorg.Chem.,2011,50:10660-10666
