不銹鋼絲網上氧化鋅納米線陣列的可控生長及光催化性能研究*
2013-10-17 08:45:34趙志超曲寶涵宋祖偉
無機鹽工業 2013年12期
趙志超,曲寶涵,宋祖偉,代 輝
(青島農業大學化學與藥學院,山東青島 266109)
光催化技術是近年來發展起來的一種新型降解有機污染物的技術,由于其具有高效、價廉、對環境友好、容易循環使用等優點,備受研究者的關注[1-2]。開發性能優異的光催化材料是光催化降解研究領域的核心課題。各種半導體氧化物如氧化鈦[3]、氧化錫[4]、氧化鋅[5-7]等被相繼開發研究。 其中,氧化鋅是一種具有較高的電子遷移率[8]、高激發能的半導體材料,且其催化效率高,成本低廉,對環境友好[9],因而被認為是極具應用前景的高活性光催化材料。目前,應用較廣泛的為氧化鋅納米粉末材料,但是納米粉末光催化材料在實際應用中存在著不易分散、易團聚、難回收再利用等問題[10-11]。因此,光催化材料的負載化對光催化技術的實用化非常重要,選擇合適的基底材料是制備負載型納米氧化鋅的核心問題。不銹鋼絲網具有良好的平面穩定性、導電性、導熱性,且其網狀結構有利于提高反應溶液的流動性、光電子的傳輸和增大納米結構的比表面積,提高光催化效率。筆者通過簡易的水熱法[12],以不銹鋼絲網為基底,制備了氧化鋅納米線陣列,借助掃描電鏡對制備的氧化鋅納米線陣列的形貌進行了表征,并分析了制備產品的光催化性能。
1 試驗部分
1.1 氧化鋅納米線陣列的制備
不銹鋼絲網的清洗:不銹鋼絲網(2.5 cm×3.0 cm,孔徑38 μm)分別用丙酮、無水乙醇超聲波清洗30 min,連續重復3次,60℃干燥箱中干燥備用。
不銹鋼絲網上氧化鋅納米線陣列的制備:把不銹鋼絲網浸入到0.005 mol/L的乙酸鋅無水乙醇溶液中約10 s,在紅外燈下快速干燥,連續重復4~5次,隨后在馬弗爐中在350℃煅燒20 min(在不銹鋼絲網上涂上了一層氧化鋅晶種層)。分別配制濃度 為 0.025、0.05、0.1、0.2 mol/L 的 硝 酸 鋅 溶 液 各40 mL,分別加入 1.0、2.0、2.5、4.0 g 濃氨水,攪拌均勻,將上述溶液倒入50 mL內襯聚四氟乙烯的水熱反應釜中,并把一片涂有氧化鋅晶種層的不銹鋼絲網貼壁豎直放入反應釜中,密封后在干燥箱中于95℃反應不同的時間。所得樣品分別用去離子水、無水乙醇洗滌3次,60℃干燥6 h。
1.2 樣品形貌及性能表征
采用JSM6700F型場發射掃描電子顯微鏡(SEM)觀察樣品的形貌。以橙黃Ⅱ的脫色降解為模型反應,光源為主波長365 nm、20 W殺菌紫外燈,溶液液面與紫外燈的距離為20 cm。將樣品平放入80 mL橙黃Ⅱ溶液中,在暗箱中進行預吸附30 min以達到吸附平衡,在不斷攪拌下開始光照,每1 h取一次樣品。試樣分析采用紫外-可見分光光度法,經掃描測知橙黃Ⅱ的最大吸收波長為486 nm,橙黃Ⅱ的濃度分析在UV-2450型紫外-可見分光光度計上進行。
2 結果與討論
2.1 形貌表征
圖1a、b分別為不銹鋼絲網(彎曲狀)上氧化鋅納米線陣列的光學照片和SEM照片。由圖1a可以看出,氧化鋅陣列宏觀上生長很均勻,表面光滑,更重要的是氧化鋅陣列不會因為基底高度彎曲而發生脫落或松散,展現出很強的附著力,從而生長其上的氧化鋅陣列在柔軟型光電器件上有著潛在的應用價值。由圖1b可以清晰地看到,氧化鋅納米線均勻、致密地生長在不銹鋼絲網上。

圖1 不銹鋼絲網(彎曲狀)上氧化鋅納米線陣列的光學照片(a)和SEM照片(b)
圖2為不同硝酸鋅濃度、反應時間分別為3 h和6 h條件下制備的氧化鋅納米線陣列的SEM照片,其陣列長度見表1。由圖2可以看出,反應時間為3 h的樣品(a~d),氧化鋅納米線的生長尚不完全,端面不平整,有明顯缺陷;反應時間為6 h時的樣品(e~g),氧化鋅納米線具有清晰的六棱柱狀外觀,端面平整,沒有缺陷,呈規則的六邊形,具有明顯的極性生長特征,但是納米線的直立性和分散性稍差[13]。結合表1,在相同硝酸鋅濃度下,反應6 h制得的氧化鋅納米線的長度大于反應3 h制得的氧化鋅納米線的長度,說明反應時間對氧化鋅納米線的晶形和長度有重要影響。

圖2 不同反應時間、不同硝酸鋅濃度下制備的氧化鋅納米線陣列SEM照片
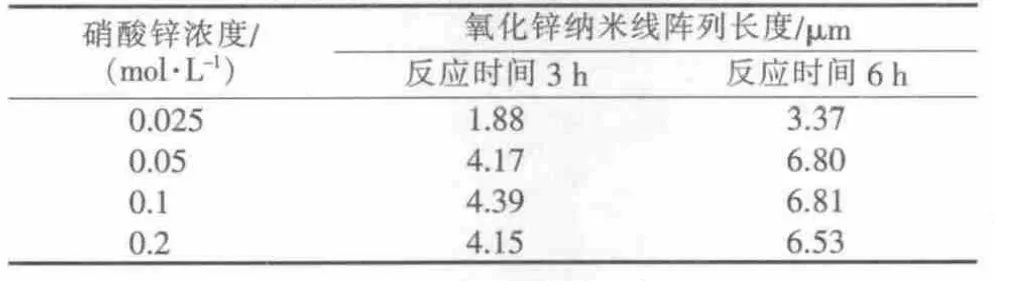
表1 不同反應時間、不同硝酸鋅濃度下制備的氧化鋅納米線陣列的長度
圖3為反應時間為6 h、不同硝酸鋅濃度下制備的氧化鋅納米線陣列SEM照片。從俯視圖(a、c、e、g)看,隨著硝酸鋅濃度的增加,氧化鋅納米線陣列的密度、直徑、產率逐漸變大,從硝酸鋅濃度為0.025 mol/L的稀疏狀態變為濃度為0.2 mol/L的致密狀態,直立性也變得越來越好,這一點從其高倍圖像圖2e與圖2h也可以明顯看出。

圖3 不同硝酸鋅濃度下(反應時間6 h)制備的氧化鋅納米線陣列SEM照片
由側視圖(b、d、f、h)并結合表 1 得知,氧化鋅納米線陣列的長度隨著硝酸鋅濃度的增加逐漸變大,其中當濃度為0.1 mol/L時納米線的長度達到最大,而當濃度為0.2 mol/L時納米線陣列的長度反而減小。由圖2g和圖3h看出,除了較大直徑的單個柱狀氧化鋅納米線以外,也有很多直徑小的柱狀氧化鋅納米線出現,而且其與較大直徑的單個柱狀組成團簇。這說明隨著硝酸鋅濃度的增大,單個柱狀的直徑先是增大,其后增長速度很慢,而新的柱狀氧化鋅不斷生長出來與原先較大的單個柱狀組成團簇[14]。另外,由圖2e~g可以看出,在硝酸鋅濃度較低時,單根氧化鋅納米線頂端和中端直徑相差很大,隨著硝酸鋅濃度的增大這種差距逐漸減小,當硝酸鋅濃度達到0.2 mol/L時幾乎沒有差別,成為規則的六棱柱狀。
2.2 光催化性能表征
圖4為以不同硝酸鋅濃度、不同反應時間制備的氧化鋅納米線陣列為光催化劑,光照橙黃Ⅱ溶液8 h后橙黃Ⅱ的剩余含量。由圖4可以看出,在相同硝酸鋅濃度下,反應6 h制備的樣品其光催化性能明顯高于反應3 h的樣品。這是由于,同等硝酸鋅濃度下反應3 h制備的氧化鋅納米線陣列的密度、產率、長徑比均小于反應6 h制備的樣品,使得光催化劑的含量和表面積均小于反應6 h的樣品。另外,反應3 h制備的氧化鋅納米線陣列結晶度較差,有過多的缺陷位置,這有可能成為光生載流子的復合中心,從而降低量子效率,而使陣列的光催化性能下降[15]。
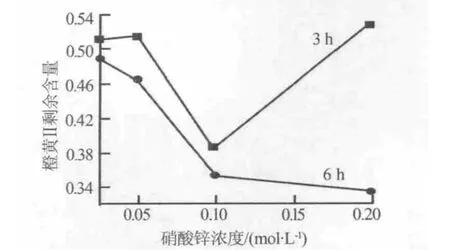
圖4 水熱反應時間對氧化鋅納米線陣列光催化性能的影響
圖5為以反應時間為6 h、不同硝酸鋅濃度下制備的氧化鋅納米線陣列為光催化劑,橙黃Ⅱ的剩余含量隨光照時間的變化曲線。從圖5看出,沒有氧化鋅納米線陣列為光催化劑的橙黃Ⅱ溶液其光催化降解率變化很小,說明氧化鋅納米線陣列具有較高的光催化活性。隨著硝酸鋅濃度的增加,氧化鋅納米線陣列的催化性能逐漸增強。但是,硝酸鋅濃度為0.2 mol/L條件制備的樣品與硝酸鋅濃度為0.1 mol/L條件制備的樣品相比,其催化性能增強的幅度極小。這是由于,隨著硝酸鋅濃度的增加,生成的氧化鋅納米線陣列的密度和厚度逐漸增大,不利于吸附更多的橙黃Ⅱ分子且透光性變差,光子吸收率降低,使得光催化性能下降[16]。結合圖4和圖5看出,硝酸鋅濃度為0.1 mol/L、反應時間為6 h條件下制備的氧化鋅納米線陳列樣品具有較優的光催化性能。
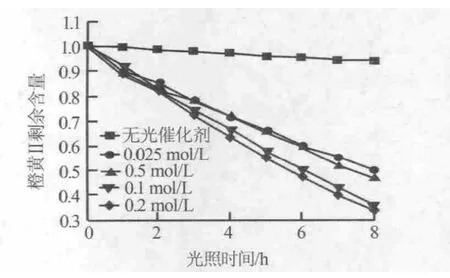
圖5 硝酸鋅濃度對氧化鋅納米線陣列光催化性能的影響
圖6為硝酸鋅濃度為0.1 mol/L、反應時間為6 h制備的氧化鋅納米線陣列在相同條件下降解橙黃Ⅱ溶液的循環性能測試。由圖6可見,4次循環曲線在任何光照時間點其光催化降解效果變化很小,說明氧化鋅納米線陣列催化性能具有較高的穩定性,可以多次重復使用,因而具有潛在的應用價值。
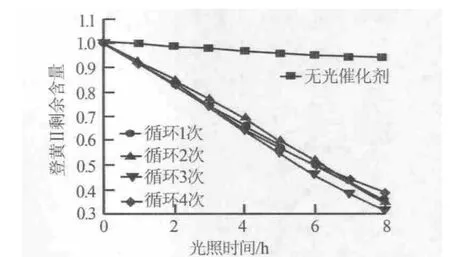
圖6 氧化鋅納米線陣列的循環性能
圖7為硝酸鋅濃度為0.1mol/L、反應時間為6 h制備的氧化鋅納米線陣列兩層疊合在一起作為光催化劑,橙黃Ⅱ剩余含量隨光照時間的變化曲線。由圖7可見,雙層疊合的氧化鋅納米線陣列的光催化性能明顯高于單層氧化鋅納米線陣列。這是由于,不銹鋼絲網具有網狀結構,其能透過光和反應溶液,第二層的氧化鋅納米線陣列同時可以接受光并產生光降解效應,這為制備多維納米陣列和薄膜提供了一種新的研究思路。
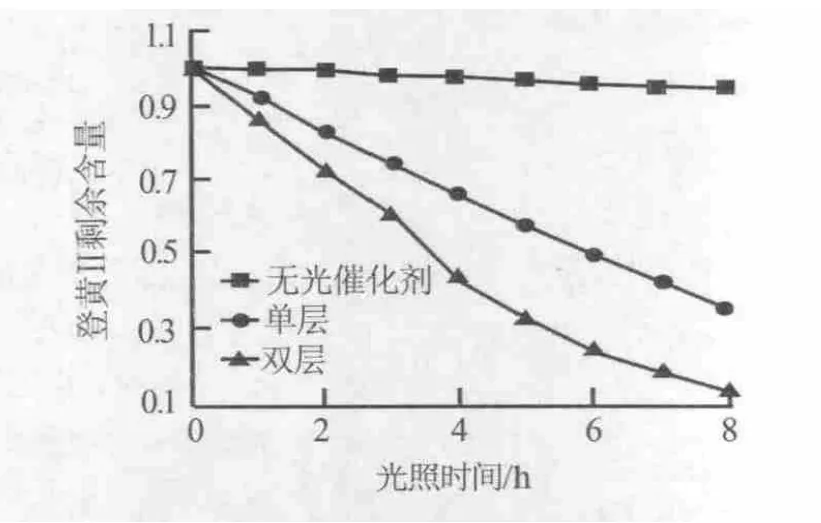
圖7 雙層氧化鋅納米線陣列光催化降解橙黃Ⅱ曲線
3 結論
以硝酸鋅和濃氨水為原料,不銹鋼絲網為基底,通過水熱法制備出氧化鋅納米線陣列。SEM表征結果表明,氧化鋅納米線陣列的密度、長度、直徑和晶形與硝酸鋅的濃度、反應時間有很大關系。以不同條件制備的氧化鋅納米線陣列為光催化劑,對橙黃Ⅱ溶液進行光催化降解實驗,發現硝酸鋅濃度為0.1 mol/L、反應時間為6 h條件下制備的氧化鋅納米線陣列具有較優的光催化降解性能,并展現出了良好的循環使用性。另外,雙層疊合氧化鋅納米線陣列的光催化降解性能優于單層氧化鋅納米線陣列的實驗結果,其不銹鋼絲網的網狀結構為制備多維納米陣列和薄膜提供了一種新的研究思路。
[1]Huang M H,Mao S,Feick H,et al.Room-temperature ultraviolet nanowire nanolasers[J].Science,2001,292:1897-1899.
[2]Wang Z L.Zinc oxide nanostructures:growth,properties and applications[J].J.Phys.:Condens.Matter,2004,16:R829-R858.
[3]Ren W J,Ai Z H,Jia F L,et al.Low temperature preparation and visible light photocatalytic activity of mesoporous carbon-doped crystalline TiO2[J].Applied Catalysis B:Environmental,2007,69(3/4):138-144.
[4]Pang G,Chen S,Koltypin Y,et al.Controlling the particle size of calcined SnO2nanocrystals[J].Nano Lett.,2001,1(12):723-726.
[5]Zhang Y,Jia H B,Yu D P.Metal-catalyst-free epitaxial growth of aligned ZnO nanowires on silicon wafers at low temperature[J].J.Phys.D:Appl.Phys.,2004,37:413-415.
[6]Liu C H,Zapien J A,Yao Y,et al.High-density,ordered ultraviolet light-emitting ZnO nanowire arrays[J].Adv.Mater.,2003,15(10):838-841.
[7]Gupta V,Bhattacharya P,Yuzuk Y I,et al.Optical phonon modes in ZnO nanorods on Si prepared by pulsed laser deposition[J].J.Cryst.Growth,2006,287(1):39-43.
[8]Yu H D,Zhang Z P,Han M Y,et al.A general low-temperature route for large-scale fabrication of highly oriented ZnO nanorod/nanotube arrays[J].J.Am.Chem.Soc.,2005,127(8):2378-2379.
[9]張艷輝,田彥文,趙迎憲,等.半導體氧化鋅的制備及其光催化性能研究[J].兵器材料科學與工程,2008,31(1):60-63.
[10]Wang Y,Li X,Lu G,et al.Synthesis and photo-catalytic degradation property of nanostructured-ZnO with different morphology[J].Mater.Lett.,2008,62(15):2359-2362.
[11]Patra M K,Manoth M,Singh V K,et al.Synthesis of stable dispersion of ZnO quantum dots in aqueous medium showing visible emission from bluish green to yellow[J].J.Lumin.,2009,129(3):320-324.
[12]余可,靳正國,劉曉新,等.氨水溶液制備ZnO納米晶陣列的研究[J].無機化學學報,2006,22(11):2065-2069.
[13]Pearton S J,Norton D P,Ip K,et al.Recent progress in processing and properties of ZnO[J].Prog.Mater Sci.,2005,50(3):293-340.
[14]Ma T,Guo M,Zhang M,et al.Density-controlled hydrothermal growth of well-aligned ZnO nanorod arrays[J].Nanotechnology,2007,18(3):035605.
[15]Wu J J,Wen H I,Tseng C H,et al.Well-aligned ZnO nanorods via hydrogen treatment of ZnO films[J].Adv.Funct.Mater.,2004,14(8):806-810.
[16]Ye Z Z,Huang J Y,Xu W Z,et al.Catalyst-free MOCVD growth of aligned ZnO nanotip arrays on silicon substrate with controlled tip shape[J].Solid State Commun.,2007,141(8):464-466.
