基于活性金電極上硫代水楊酸自組裝單分子層的電化學表面增強拉曼光譜
2013-10-18 05:27:34劉文涵袁榮輝滕淵潔馬淳安
物理化學學報 2013年12期
關鍵詞:振動
劉文涵 袁榮輝 滕淵潔 馬淳安
(浙江工業大學化學工程與材料學院,綠色化學合成技術國家重點實驗室培育基地,杭州 310032)
1 引言
表面增強拉曼散射(SERS)光譜憑借其極高的檢測靈敏度,在表面結構、表面吸附以及界面特性等領域1-3得到了廣泛的運用,已使其成為在分子水平上表征金屬/溶液界面的電化學過程最為有效的技術.
分子自組裝技術可在基底表面形成穩定有序、可控、均一整齊的自組裝單分子層(SAMs),已廣泛應用于制備功能性薄膜、分子識別、功能化修飾以及電極表面改性等研究中,特別是硫醇化合物的SAMs研究較為突出.4-6含巰基的分子在金表面成膜已被應用于傳感器、修飾電極等研究領域,7,8當其吸附到金電極表面時,可以形成穩定的單分子層,使得修飾電極呈現出新的電化學特性.9,10硫醇類化合物在經過特殊處理的金表面會產生很強的特征拉曼光譜信號.11-13硫代水楊酸(TSA)為芳香族硫醇化合物,芳基硫醇分子具有高度的各向異性,且TSA較其他對位的巰基化合物制備容易、成本較低.以活性金作基底材料相比活性銀不容易在空氣中形成氧化膜,也不易被電腐蝕.因此,對于金表面的TSA自組裝單層的研究具有一定的理論意義和潛在的應用前景.
本文采用電化學活化金電極表面的方法,制備具有表面活性的粗糙金電極,利用原位電化學表面增強拉曼光譜(EC-SERS)技術,在分子水平上表征電吸附在金表面的TSA單分子層,研究了苯環上帶有的硫醇端基的振動光譜特性.探討了不同電位等條件下對TSA溶液EC-SERS的影響.并通過計算TSA在不同pH條件下存在形式的分布以及施加高負電位前后的增強因子(EF),進而研究了TSA在金電極上的電化學吸附取向并對機理進行了探討.
2 實驗部分
2.1 儀器與試劑
LabRAM HR UV 800激光顯微拉曼光譜儀(法國JOBIN YVON公司);激發光源:632.81 nm He-Ne激光器;共焦孔徑300 μm,光柵刻線數600 lines·mm-1,物鏡:50倍長焦距鏡頭.CHI660B型電化學工作站(上海辰華儀器有限公司).PHSJ-4A型實驗室pH計(上海精密科學儀器有限公司).直徑3 mm金盤電極(天津艾達恒晟科技發展有限公司).
硫代水楊酸(99%,Aladdin試劑有限公司),其它試劑均為分析純及以上.粒徑30-50 nm的氧化鋁拋光粉,水為18.3 MΩ·cm超純水,由HUMAN UP900型超純水器(韓國HUMAN公司)制得.
2.2 金電極電化學活化
未“活化”的金表面不能產生SERS效應,需先進行粗糙活化處理:先將金圓盤電極在金相砂紙上打磨,再將細Al2O3拋光粉與水混合成懸浮液,在定性濾紙上將電極拋成鏡面,依次用無水乙醇、超純水超聲各2 min,獲得潔凈的電極表面.以金盤電極為工作電極,鉑絲為輔助電極,飽和甘汞電極(SCE)為參比電極,組成三電極體系.0.3 mol·L-1硫酸為支持電解質底液,以100 mV·s-1的掃描速率在-0.2 V至上限1.6 V的電位區間內反復進行電位掃描,以增加電極表面的活化位點,14待循環伏安特征穩定后,換用0.1 mol·L-1KCl作電解質底液,在電位區間-0.1-1.3 V范圍內,以10 mV·s-1掃描速率循環掃描3周,再用超純水清洗電極.金盤電極經電化學氧化還原(ORC)處理,獲得一層暗色且具有SERS活性的表面.
2.3 激光拉曼光譜和SERS光譜測定
常規拉曼光譜(NRS)測定:將經無水乙醇溶解并重結晶后的固體樣品置于載玻片上,于激光顯微拉曼光譜儀載物臺的物鏡視野下,通過計算機的影像窗口調整焦距,使激光聚焦于樣品表面,進行NRS測試.掃描范圍200-2800 cm-1,光譜信號采集時間10 s,積分2次平均.
液相現場單分子層SERS測定:以活性金電極作為基底材料,于光譜電化學池內,在不施加電位的情況下,電極表面在1×10-3mol·L-1、pH=3.2的TSA待測溶液中浸置90 min進行自組裝,并控制電極表面的液面厚度在1 mm內進行SERS光譜掃描.掃描范圍200-2800 cm-1,采集時間50 s,積分2次平均.
浸飾膜SERS測定:配制不同pH值的1×10-3mol·L-1TSA溶液,將活性金電極浸入不同pH值的TSA溶液中,浸置90 min進行自組裝,取出電極在高純N2氛圍中干燥,使TSA充分吸附附著在電極表面,進行修飾膜的SERS光譜檢測,掃描范圍200-1700 cm-1,采集時間10 s,積分2次平均,在獲取SERS峰強時,需測定電極表面3個不同位置點的數值取平均,峰強度數據由軟件Labspec讀出.
2.4 現場EC-SERS測定
因酸性體系TSA得到的SERS效應強于堿性體系,TSA在pH<3時有形成懸濁液而未能很好測得.故選取以0.1 mol·L-1KCl作底液,TSA濃度為1×10-3mol·L-1、pH=3.2的溶液置于光譜電化學池內,控制其電極表面的液面厚度在1 mm內,連接三電極體系,使激光聚焦在電極表面,進行原位EC-SERS測試,測定不同通電富集作用時間以及不同電位下TSA分子的EC-SERS.掃描范圍200-1700 cm-1,采集時間50 s,積分2次平均.
3 結果與討論
3.1 TSA在活性Au表面的化學吸附
為考察TSA在活性Au上的吸附作用,測定了1×10-3mol·L-1TSA溶液、pH=3.2的SERS(圖1(b))與TSA晶體的NRS(圖1(a))對比.用激光拉曼光譜直接測定1×10-3mol·L-1TSA溶液,由于濃度太低而溶液的NRS信號很不明顯,因而在1×10-3mol·L-1TSA溶液中的Au表面測得的拉曼光譜,可認為是由于TSA分子吸附鍵合在Au表面后產生的SERS光譜.非晶形粉末TSA經重結晶后的NRS以及TSA在活性Au電極上的SERS譜峰歸屬列于表1.
從圖1可以看出,Au表面的SERS相比TSA晶體的NRS發生一些變化,TSA原有的S―H特征搖擺振動弱峰(311 cm-1)消失,替代并新產生了S―Au峰(269 cm-1),15,16同時歸屬S―H伸縮振動的強峰(2520 cm-1)亦消失.SERS出現了多個新峰,NRS中共有峰的拉曼位移在SERS中出現了普遍向低波數移動的現象(表1).表明TSA已在“粗糙”的Au表面發生了化學吸附并促進了拉曼的化學增強等作用.

圖1 TSA晶體NRS(a)與1×10-3 mol·L-1 TSA溶液在活性Au表面的SERS光譜(b)Fig.1 Normal Raman spectroscopy(NRS)of TSAcrystal sample(a)and SERS spectrum of 1×10-3 mol·L-1 TSA solution on activated Au surface(b)
3.2 不同pH值下的浸飾TSA單層膜的SERS光譜
將活性Au電極浸入不同pH值的TSA溶液中,靜置90 min進行自組裝,再取出電極在高純氮中干燥,使TSA充分吸附附著在Au表面形成浸飾膜,進行SERS測試,結果見圖2A.
當測試Au表面上不同pH的TSA時,均可觀察到S―Au的伸縮振動峰,表明TSA通過S―H失去質子與Au形成強的S―Au鍵吸附于電極表面成“膜”,隨著pH的上升,苯環的呼吸特征峰(1035 cm-1)強度逐漸變弱;其它基團的特征峰,如453 cm-1的雙取代苯振動、481 cm-1的C―S面外彎曲振動、647 cm-1的1,2-雙取代苯骨架振動以及697 cm-1歸屬C―S伸縮振動等峰高亦隨之降低.
以最強峰苯的環呼吸特征峰強與pH作關系圖(圖2B).可以看出,隨著pH增加,SERS的峰強信號逐漸減弱,其變化趨勢以pH 5-6為分界,表現為不同的2個階段,在pH 6及以上時峰強I2隨著pH的變化的下降速率即斜率為pH 3-5時I1的2倍,表明SERS效應下降速率增加了1倍.當pH>11時譜峰強度已很弱,表明堿性體系已不利于TSA在Au界面上的成鍵結合或不利于增強效應的產生,表觀結果是OH-的存在會加速阻止TSA的SERS效應.

圖2 不同pH值對TSA修飾膜的SERS光譜(A)及苯環呼吸特征峰強(B)的影響Fig.2 SERS spectra of gold electrodes modified with TSA(A)and intensity of 1035 cm-1 SERS peak at different pH values(B)
3.3 TSA在活性Au表面的EC-SERS
為了進一步考察Au電極表面的TSA在通電條件下的吸附性能,及TSA與Au基底在不同的恒電位條件下的相互作用,分別探討了不同電富集時間以及不同電位條件對EC-SERS的影響.
由于電化學ORC得到的粗糙Au表面,在較高電位下仍然會發生電化學溶出現象,粗糙Au原子變為Au離子而失去SERS效應.因而需先考察其電極行為,以探討合適的電化學吸附所需的電位.電極經拋光、超聲清洗、活化處理,將Au工作電極浸入0.1 mol·L-1KCl溶液中,使用三電極體系對自組裝TSA修飾膜后的Au電極進行陽極極化曲線測試,掃描速率為10 mV·s-1.按同一方法測定未經修飾的活性Au工作電極在0.1 mol·L-1KCl底液的陽極溶出電位作為本底對比,電化學實驗均在室溫下進行((23±2)°C).結果表明,電化學溶出腐蝕電位均在0.8 V以上.為保證ORC活化后的金基底的ECSERS活性和同時作為工作電極而不被電氧化,其所施加的正電位不應超過0.8 V.
3.3.1 不同電富集時間對EC-SERS的影響
將活性Au電極浸沒于以0.1 mol·L-1KCl作支持電解質底液,TSA為1×10-3mol·L-1、pH=3.2的待測液中.測定了0.1 V電位下,在10-100 s(間隔10 s)的不同電預富集時間的EC-SERS,結果如圖3A.并以苯的環呼吸特征峰強與電富集時間作關系圖(圖3B).
可看出,在一定條件下,通電吸附累積到70 s時,峰強最大,繼續延長時間,EC-SERS效應反而下降.推測其可能的機理是隨著電富集時間的延長,Au電極表面吸附的TSA分子或分子層逐漸增多,反而阻止了EC-SERS作用,表明在1×10-3mol·L-1數量級時,電富集70 s下TSA組裝形成SAMs有最佳的效果.另外可能,S―H與Au表面形成S―Au鍵合后,C6H4(COOH)SH團簇向外層空間伸展,隨著電累積時間的增加,分子內及分子間的彼此接近,產生“擁堵”而引起空間阻礙,形成分子的空間位阻效應,從而減弱了其吸收激發光能量的同時阻礙了振動所散射出的拉曼信號,因而EC-SERS信號減弱.

表1 TSA晶體的NRS,TSA在金電極上的SERS與EC-SERS位移(cm-1)對比及譜峰歸屬15,17,18Table 1 Comparison of Raman frequencies(cm-1),intensities and vibrational modes of TSAin NRS of crystal,SERS,and EC-SERS15,17,18

圖3 0.1 V電位(vs SCE)下不同電富集時間對TSA的EC-SERS(A)及苯環呼吸特征峰強(B)的影響Fig.3 EC-SERS of TSA(A)and intensity of 1036 cm-1 peak with different accumulation time at 0.1 V(vs SCE)(B)
3.3.2 不同電位與EC-SERS的關系
測定了在不同的恒電位條件下,以0.1 mol·L-1KCl為底液,1×10-3mol·L-1、pH 3.2的TSA溶液,在電化學預富集70 s時間下的EC-SERS,見圖4.TSA在Au電極上的SERS以及EC-SERS位移對比和具體譜峰歸屬列于表1.從表1的對比中可以看出,采用單純溶液富集與電化學富集組裝,S―Au的伸縮振動向高波數發生了移動(269→271 cm-1),同時增加了一些特征譜峰,共有的譜峰有所增強(w→m,m→s,s→vs),且基本都發生了右移,表明Au表面施加的電場,在活性Au吸附C6H4(COOH)SH時起到了一定的電化學增強的作用.

圖4 不同電位條件下金電極表面TSA單分子層的EC-SERSFig.4 EC-SERS of TSAmonolayer on gold electrode at different potentials from+0.7 to-1.4 V(vs SCE)
同時考察了在電位0.2 V下,通電預富集70 s再斷開電路后,測定10、200、300、500 s時的EC-SERS信號,結果表明譜峰信號及位移的變化不大,僅峰強略有增加.
3.4 TSA在不同pH值條件下的存在形式及機理探討
TSA(C7H6O2S)在水中存在著離解平衡,其p Ka1值19為4.05,在水中不存在p Ka2使C7H5O2S-繼續離解為C7H4O2S2-.圖5是根據酸堿常數,計算與繪制TSA在不同pH值時的形態分布.δ1為中性分子C6H4(COOH)SH的分布分數,δ2為負一價離子C6H4(COO-)SH的分布分數(δ2=1-δ1).TSA為弱酸,當pH在3以下時,基本以中性分子形式存在,解釋了TSA在pH<3時有形成懸濁液的現象.pH 4.05時中性分子與負一價離子的數目相等;隨著pH升高,δ1含量逐漸減少,當pH升至6時,中性分子數目減少至1/100,此后基本以δ2為主.
3.4.1 TSA在Au表面的化學吸附機理
結合圖2、5可以看出,溶液的酸堿度對TSA在Au表面的SERS光譜有一定的影響,隨著pH值及TSA分子狀態的改變,基本可分2個階段.
溶液pH在5以下時,酸度較大,氫離子數目較多,羧基數目還較多,COOH中C=O、C―O誘導吸/拉電子能力較強(圖6(a)),故S的電子云密度減小,對H的束縛力減弱,容易失去H與活性Au作用成鍵,與基底Au形成“強烈”吸附,自組裝成單層膜而產生SERS且信號較強.
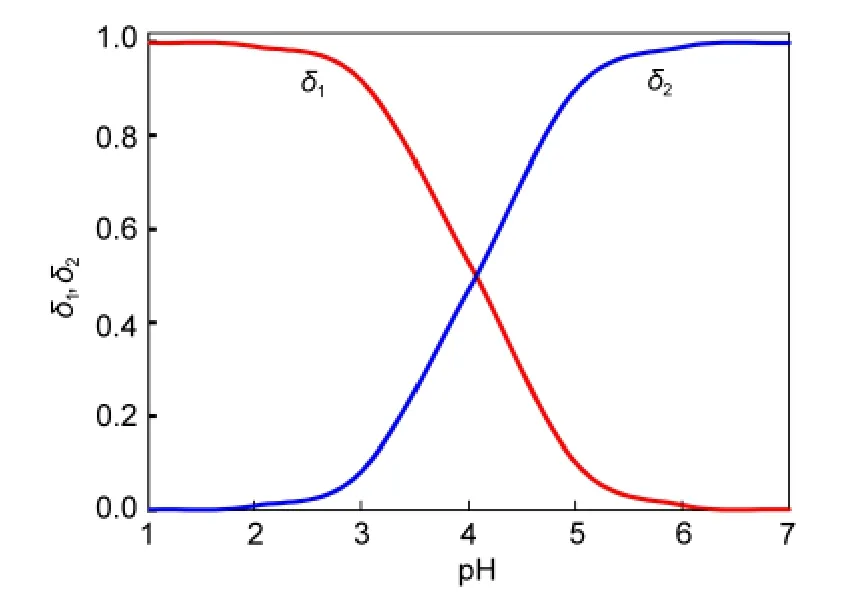
圖5 TSA的分布分數(δ1,δ2)與溶液pH的關系Fig.5 Distribution fraction(δ1,δ2)of TSA at different pH values
當溶液逐漸趨向堿性時,TSA發生解離,在pH 6以上時,基本以負一價δ2存在,所產生的COO-有很強的供電子能力,苯環和羰基π-π共軛產生給電子作用,使S原子吸引H的能力增強(圖6(b)),使得S―H中的H不易脫去,造成S―Au較難成鍵,因而信號減小.隨著溶液中OH-的增加,SERS峰強發生了線性的下降,此時是否OH-也參與了競爭吸附,而排斥了TSA在Au表面的吸附,抑或是強堿性條件下,大量產生的COO-酸根與活性Au部分成鍵,使TSA的吸附方式發生了改變,垂直吸附(見圖7(a))轉化為“斜躺”式(圖7(c)),而造成SERS效應減弱.
3.4.2 TSA在Au電極表面的電化學吸附機理及增強因子計算
在pH 3.2對1×10-3mol·L-1TSA溶液進行ECSERS測定時,其溶液中有δ1和δ2兩種形態,且δ1的含量占87.6%,而δ2只占12.4%;故pH 3.2的TSA在Au上起到界面相互作用的主要形態是δ1.

圖6 TSA在不同酸堿下的化學吸附作用機理Fig.6 Chemical absorption mechanisms of TSA monolayer at acid(a)and base(b)conditions
從圖4可以看出,峰強度隨著電位正移而增大,0.7 V時EC-SERS信號最強.根據SERS表面選擇定律,20,21直接作用于或靠近SERS基底的分子或基團,其在垂直方向上有最大分量的振動模式可得到最大增強,且越垂直于SERS基底的振動,增強效應越明顯.因此在所加電位不產生電化學溶出基底材料的前提下,S原子已與基底Au發生了一定的鍵合作用,且在外加電位越正時,振動增強作用越強,越有效;或亦表明電位越正,在電場的作用下垂直方向排列的分子比例越多,因而歸屬S―Au伸縮振動的271 cm-1峰強和相應的其它EC-SERS峰越能明顯地呈現出來.
EC-SERS與SERS相互對比,可以看出施加電位≥0.4 V時,EC-SERS多出了歸屬苯C―H面內變形的譜峰(1180和1372 cm-1)(表1),此外歸屬C=C伸縮振動的譜峰(1534 cm-1)也被檢測到;同時苯C―H面內彎曲(1121,1164 cm-1)峰的強度顯著增大(圖4);而分子面內和面外振動的峰強度比值越大,即面內振動峰相對較強時,也說明了分子越垂直于基底.另外EC-SERS中歸屬S―H伸縮振動峰2520 cm-1沒有出現(表1),同樣認為溶液本底中的δ1通過硫醇端基失去質子,與Au形成S―Au吸附鍵合產生了EC-SERS,其垂直吸附作用于帶正電的Au電極表面(圖7(a)),而羧基向外暴露.而1255 cm-1歸屬C―O伸縮振動的譜峰很弱,可推斷COOH與Au基底有一些距離.
此外,苯環上帶負電荷的COO-也可作為另一個作用位點接近帶正電的Au電極表面形成“靜電吸附”(圖7(b),圖7(c)).從而認為少量的δ2是通過巰基和羧酸根COO-以較斜躺的方式自組裝在電化學活化的帶正電的Au表面.

圖7 金電極表面TSA在正電位作用下的吸附取向Fig.7 Proposed orientations of TSAadsorbed on gold electrode at positive potential
另一方面,由于在負電位時,帶負電的Au會與COO-相互排斥,而溶液中δ2約為12.4%,所產生的信號基本可以忽略,只需考慮占大多數的δ1形態在帶負電位的電極表面的相互作用.從圖4中EC-SERS信號強度是隨負電位大小的變化而改變,可知吸附取向也與負電位值有直接的影響關系.當施加電位負移時,最強峰苯環呼吸振動的強度逐漸減弱,推測是通過S原子吸附在Au表面的TSA分子中的苯環由垂直方式逐漸趨于傾斜.同時與苯環相關的譜峰,如805 cm-1歸屬取代苯C―H面外彎曲、C―C伸縮振動(1432,1463 cm-1)和苯衍生物的環伸縮雙峰(1560,1585 cm-1)等峰強都逐漸減弱,表明苯環有逐漸脫離基底表面的趨勢.由于在此濃度下,常規拉曼光譜檢測不到拉曼信號,所產生的信號均由SERS產生,因而TSA分子的拉曼光譜均會隨分子吸附的改變而發生變化.當外加電位至-1.0 V時,信號繼續變弱,且峰強已很不明顯了,推測δ1在帶負電的Au上開始部分脫離或發生吸附取向的變化.從伏安掃描圖中表明,S―Au的還原峰起始電位出現在-0.91 V,峰值在-1.07 V左右.當電位進一步負移至-1.4 V時,EC-SERS譜峰基本上消失,故可推斷活性Au電極表面的TSA,在高負電位下發生了還原脫附.22,18,23其吸附取向隨負電位增大的變化過程如圖8所示.
同時,經電化學粗糙化處理后的電極,其表面具有亞穩態納米結構,基于EF與電位的密切相關性,24不同電位對EF的影響同樣值得關注.
本文選用垂直吸附取向的TSA(pH=3.2)作為探針分子來進行活性Au表面的EF估算,EF的求取方法25-27見式(1):

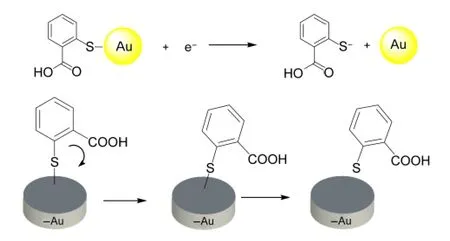
圖8 金電極表面TSA的吸附取向隨負電位增大的變化過程Fig.8 Orientation change of TSAmonolayer covered on gold electrode with the increase of negative potential
其中,Isurf為吸附在基底上TSA分子的SERS特征峰強,Nsurf表示活性Au基底上激光所照射到的TSA分子數,Ibulk為溶液中TSA的NRS特征峰強,Nbulk表示溶液中NRS測試時激光所照射的有效TSA探針分子數.此處,Isurf和Ibulk可分別從1×10-3mol·L-1TSA的SERS及0.5 mol·L-1TSA本體溶液的NRS譜圖中,對應選擇苯環的呼吸特征峰強直接得到(Ibulk=751.63).而Nsurf和Nbulk需通過計算,結合3.3.1節,判斷浸置90 min、電預富集70 s進行自組裝的1×10-3mol·L-1TSA分子為飽和吸附,則Nsurf可由式(2)表達:

σ為單個吸附分子占的表面積,金基底上的σ值為常數28(σAu=0.27 nm2).A為激光聚焦焦點的有效面積.R為活性Au基底的粗糙因子,可采用Trasatti和Petrii29的方法計算,通過比較Au在0.3 mol·L-1硫酸底液中,ORC活化前表面平整的基底上Au3+還原峰電量Q(對應Au3+還原曲線的積分面積)以及活化后粗糙表面的Au3+還原峰電量(Q*)可得,如(3)式:

A*為ORC活化后工作電極的有效面積,Ar為直徑3 mm金盤電極的幾何面積,求得R=1.104.
式(1)中的Nbulk需在考慮共焦顯微拉曼的共焦特征的基礎上進行計算,30見式(4):

C*為TSA本體溶液的濃度(C*=0.5 mol·L-1),NA為阿伏伽德羅常數(NA=6.02×1023mol-1).h為激光束照射到溶液中理想聚焦面的有效深度,可按álvarez-Puebla等31的方法來計算,如式(5)所示:

n為環境媒介反射因子,即波長λ=632.81 nm的激光從空氣介質到溶液中的折射率(n=4/3),N.A.為50倍長焦距物鏡的數值孔徑(N.A.=0.55),通過計算得到本文所用激光顯微拉曼光譜儀的h=2.789 μm.
因0.5 mol·L-1TSA本體溶液NRS的光譜信號采集時間為80 s,在對本體溶液不同的采集時間進行折算校正的基礎上,綜合式(1)、(2)、(4)進一步推導可得:


圖9 電位條件從正到負過程中活性金電極上TSA單分子層的吸附與還原脫附過程Fig.9 Adsorption and reductive desorption process of TSAmonolayer on activated gold electrode potential varying from positive to negative
則1×10-3mol·L-1TSA在采集時間50 s的條件下,由式(6)最后求得施加0 V時的EF0=4.74×103,0.4 V下的EF0.4=8.26×103,-1.4 V下的EF-1.4=319,施加電位-1.8 V之后再回至0.4 V的EF-1.8/0.4=137,說明加了高負電位后再加正電位,其EF仍然較小,表明電極的拉曼增強活性已降低.
結果表明,SERS基底在高負電位的作用下,EF明顯減小,TSA發生了還原脫附,此時,因電位再回復至正電位,EF值仍較小,表現出電極性能已發生了改變,并不可逆地降低或失去了拉曼光譜增強的活性.TSA單分子層在電位條件從正到負過程中的吸附與還原脫附過程見圖9.
4 結論
在經電化學ORC粗糙活化處理后的Au電極表面,TSA通過斷裂S―H鍵與Au形成S―Au鍵而吸附,在活性Au表面進行自組裝形成SAMs,并產生SERS以及EC-SERS.在酸性介質中,由于COOH中C=O、C―O誘導吸/拉電子的作用,比在堿性條件下更易自組裝成單層膜,而產生較強的SERS效應.在現場EC-SERS中,加0.7 V和電預富集70 s下的信號最強,隨著電位負移,尤其在還原電位后,EF不可逆的減小,信號逐漸減弱,直至基本消失.表明TSA在Au表面的自組裝過程的SERS信號受到電位和吸附時間的影響,TSA分子在Au表面排布狀態會隨施加條件的改變而發生變化.在-1.4 V時發生了不可逆的還原/解吸,同時亦造成Au表面活性的降低,使EC-SERS信號消失.另外作者還推測,是否單分子層的排布、電磁場的物理抑制、電荷轉移機制等其它原因,也造成了一定的影響?這些都有待進一步計算和探討.
(1)Titus,E.J.;Weber,M.L.;Stranahan,S.M.;Willets,K.A.Nano Lett.2012,12(10),5103.doi:10.1021/nl3017779
(2)Su,Q.Q.;Ma,X.Y.;Dong,J.;Jiang,C.Y.;Qian,W.P.ACS Appl.Mater.Interfaces 2011,3(6),1873.doi:10.1021/am200057f
(3)Freye,C.E.;Crane,N.A.;Kirchner,T.B.;Sepaniak,M.J.Anal.Chem.2013,85(8),3991.doi:10.1021/ac303710q
(4)Wang,Y.Q.;Yan,B.;Chen,L.X.Chem.Rev.2013,113(3),1391.doi:10.1021/cr300120g
(5)Chu,H.;Yang,H.F.;Huan,S.Y.;Shen,G.L.;Yu,R.Q.J.Phys.Chem.B 2006,110(11),5490.doi:10.1021/jp053914m
(6)Kho,K.W.;Dinish,U.S.;Kumar,A.;Olivo,M.ACS Nano 2012,6(6),4892.doi:10.1021/nn300352b
(7)Monnell,J.D.;Stapleton,J.J.;Dirk,S.M.;Reinerth,W.A.;Tour,J.M.;Allara,D.L.;Weiss,P.S.J.Phys.Chem.B 2005,109(43),20343.doi:10.1021/jp044186q
(8)Sumner,J.J.;Creager,S.E.J.Phys.Chem.B 2001,105(37),8739.doi:10.1021/jp011229j
(9)Napper,A.M.;Liu,H.Y.;Waldeck,D.H.J.Phys.Chem.B 2001,105(32),7699.doi:10.1021/jp0105140
(10)Bertin,P.A.;Ahrens,M.J.;Bhavsar,K.;Georganopoulou,D.;Wunder,M.;Blackburn,G.F.;Meade,T.Org.Lett.2010,12(15),3372.doi:10.1021/ol101180r
(11)Ameer,F.S.;Hu,W.F.;Ansar,S.M.;Siriwardana,K.;Collier,W.E.;Zou,S.L.;Zhang,D.M.J.Phys.Chem.C 2013,117(7),3484.
(12)Dasary,S.S.R.;Singh,A.K.;Senapati,D.;Yu,H.T.;Ray,P.C.J.Am.Chem.Soc.2009,131(38),13806.doi:10.1021/ja905134d
(13)Xie,W.;Walkenfort,B.;Schlu cker,S.J.Am.Chem.Soc.2013,135(5),1657.doi:10.1021/ja309074a
(14)Yang,Y.C.;Xia,Y.;Huang,W.;Zheng,J.F.;Li,Z.L.J.Solid State Electrochem.2012,16(4),1733.doi:10.1007/s10008-011-1600-8
(15)Ohta,N.;Yagi,I.J.Phys.Chem.C 2008,112(45),17603.doi:10.1021/jp806599r
(16)Carron,K.T.;Hurley,L.G.J.Phys.Chem.1991,95(24),9979.doi:10.1021/j100177a068
(17)Ke,Y.K.;Dong,H.R.Handbook of Analytical Chemistry:Spectrial Analysis;Chemical Industry Press:Beijing,1998;pp 1153-1160.[柯以侃,董慧茹.分析化學手冊:光譜分析.北京:化學工業出版社,1998:1153-1160.]
(18)Schalnat,M.C.;Pemberton,J.E.Langmuir 2010,26(14),11862.doi:10.1021/la1010314
(19)Dean,J.A.Lange?s Handbook of Chemistry;Science Press:Beijing,1991;p 59;translated by Shang,J.F.,Cao,S.J.,Xin,W.M.[Dean,J.A.蘭氏化學手冊.尚久方,操時杰,辛無名,譯.北京:科學出版社,1991:59.]
(20)Gao,X.P.;Davies,J.P.;Weaver,M.J.J.Phys.Chem.1990,94(17),6858.doi:10.1021/j100380a059
(21)Moskovits,M.;Suh,J.S.J.Phys.Chem.1988,92(22),6327.doi:10.1021/j100333a030
(22)Kolega,R.R.;Schlenoff,J.B.Langmuir 1998,14(19),5469.doi:10.1021/la980553b
(23)Ji,W.;Kitahama,Y.;Han,X.X.;Xue,X.X.;Ozaki,Y.;Zhao,B.J.Phys.Chem.C 2012,116(46),24829.doi:10.1021/jp308805n
(24)Shafer-Peltier,K.E.;Haynes,C.L.;Glucksberg,M.R.;Van Duyne,R.P.J.Am.Chem.Soc.2003,125(2),588.doi:10.1021/ja028255v
(25)Amaya,R.O.;Rappoport,D.;Munoz,P.A.;Peng,P.;Mazur,E.;Guzik,A.A.J.Phys.Chem.C 2012,116(29),15568.doi:10.1021/jp302597v
(26)Le-Ru,E.C.;Blackie,E.;Meyer,M.;Etchegoin,P.G.J.Phys.Chem.C 2007,111(37),13794.doi:10.1021/jp0687908
(27)Jia,H.Y.Synthesis,Characterization of SERS Active Nanoparticles.Ph.D.Dissertation,Jilin University,Changchun,2006.[賈慧穎.銀納米粒子的制備、表征及其表面增強拉曼散射活性研究[D].長春:吉林大學,2006.]
(28)Stolberg,L.;Lipkowski,J.;Irish,D.E.J.Electroanal.Chem.1990,296,171.doi:10.1016/0022-0728(90)87241-B
(29)Trasatti,S.;Petrii,O.A.Pure&Appl.Chem.1991,63(5),711.doi:10.1351/pac199163050711
(30)Cai,W.B.;Ren,B.;Li,X.Q.;She,C.X.;Liu,F.M.;Cai,X.W.;Tian,Z.Q.Surf.Sci.1998,406,9.
(31)álvarez-Puebla,R.A.J.Phys.Chem.Lett.2012,3(7),857.doi:10.1021/jz201625j
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
大電機技術(2022年5期)2022-11-17 08:12:48
天天愛科學(2020年6期)2020-09-10 07:22:44
瘋狂英語·新讀寫(2020年3期)2020-06-06 09:05:56
數學物理學報(2018年4期)2018-09-14 03:40:58
數學物理學報(2017年6期)2018-01-22 02:26:40
船海工程(2015年4期)2016-01-05 15:53:26
噪聲與振動控制(2015年4期)2015-01-01 07:08:44
計算物理(2014年2期)2014-03-11 17:01:44
鄭州大學學報(理學版)(2014年3期)2014-03-01 04:21:00
