新型藏藥愈心安神滴丸的制備工藝研究
2013-11-29 08:01:02馬文俊李寶文東知多杰
中國民族醫藥雜志 2013年7期
馬文俊 李寶文 賈 明 東知多杰
(青海阿如拉藏醫藥研究開發有限公司,青海 西寧 810003)
本項目主要目標是研制一種治療和預防由氣滯血瘀所引起的疾病的藏藥新產品愈心安神滴丸,其核心研究內容主要是藥物的功效、安全性以及制劑的劑型。制備關鍵工藝技術主要是原藥材的加工炮制技術、揮發油提取工藝技術、揮發油包合工藝技術及滴丸的成型工藝技術;確定生產工藝的穩定性、可操作性及質量控制要求。
1 材料、設備
1.1 處方組成:廣棗3000g,當歸150g,干姜150g,肉豆蔻150g,訶子 150g,寒水石 150g。
1.2 主要實驗設備和檢驗儀器:磁力攪拌器、高速粉碎機、滴丸機。
1.3 制法概述:以上六味,除寒水石外,其余藥材提取揮發油,用β-環糊精包合,備用;藥渣加水煎煮三次,濾過,合并濾液,濾液濃縮成相對密度為1.26~1.28(50℃)的清膏,加乙醇使含醇量達65%,攪勻,靜置24h,取上清液,回收乙醇,濃縮干燥,粉碎并過100目篩,備用;寒水石研細并過200目篩。取聚乙二醇4000與聚乙二醇6000適量,加熱使溶融,加入上述揮發油包合物、稠膏和寒水石細粉,混勻,滴入冷卻的液體石蠟中,制成1000g滴丸,即得。
1.4 工藝路線設計[1-2]
1.4.1 處方中主要有廣棗、當歸、干姜、肉豆蔻、訶子和寒水石六味藥材,鑒于其中許多藥材含有揮發油,因此需要先提取揮發油,采用直接水浸泡后煮沸提取,冷凝收取揮發油。
1.4.2 方中寒水石為礦物類藥材,配方時需要進行奶制,主要成分為碳酸及硫酸鈣,在水中基本不溶,不宜采用提取方法。因此選擇粉碎處理后直接加入滴丸成型過程的方法;其余均為植物性藥材的果實類和根類,適合用提取的方法。
1.4.3 因原方為口服散劑,療效確定,所以提取方法采用水煮提,水提取后的提取物中有比較多的雜質(多糖、鞣質等),因此提取濃縮后應進行乙醇精制處理,以改善外觀和減少基質用量,達到外觀適當和減少用量的目的。
1.4.4 揮發油處理:由于滴丸制備時需要在比較高的溫度下進行,直接加入時揮發性成分容易損失,所以應采用保護技術,設計采用β-環糊精包合方式,在成型時以固體粉末形式和不提取的寒水石一起直接加入滴丸成型環節。
1.4.5 根據資料檢索得知,聚乙二醇是水溶性人體不吸收的常用基質,是目前在制劑中使用比較多的滴丸基質,初步選擇聚乙二醇(PEG)為首選基質。資料表明采用PEG4000和PEG6000加工的滴丸比較理想,本試驗同樣選擇上述基質進行研究,具體配合比例及用量通過實驗確定。
1.5 愈心安神滴丸工藝流程圖:
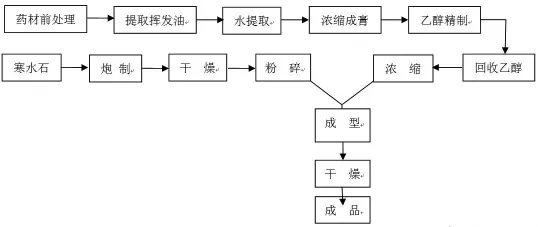
2 關鍵工藝技術實驗研究方法
2.1 劑型選擇:愈心安神滴丸是在尚未完全了解組方有效成分前提下開展的科研項目,由于復方制劑有效成分的研究仍然存在困難,提取過程中代表性成分的選擇不易確定,在保留傳統藏藥特點的基礎上結合現代藥品劑型的發展要求,選用滴丸為最終制劑劑型。
2.2 揮發油提取工藝的篩選和確定:基于研究中藥藥性、提高藥物療效、改進制劑等目的,需要將有效成分從藥材中提取分離出來。
為提高提取效率,需要對影響“傳質”的因素進行認真考慮,這些因素包括藥材的浸泡時間、提取的溶劑量、提取時間等。一般情況下,溶劑量越大,有效成分的提取越完全,但是溶劑量過大,產品的能耗增加,因此應選擇合適的提取溶劑量,使藥物的有效成分剛好能完全提取出。延長提取時間會使有效成分的提取充分,但在有些情況下,長時間高溫浸提會增加對提取物破壞的可能性。
2.2.1 提取工藝設計參考有關資料和預實驗,以水蒸氣蒸餾法提取,選加水倍量(A)、浸泡時間(B)、提取時間(C)作為考察因素,設計3個水平,以揮發油量為考察指標,設計L9(34)正交實驗。

表1 揮發油提取工藝因素水平表
2.2.2 實驗方法
根據實驗設計,選用L9(34)正交表安排實驗。每次實驗分別稱取除寒水石外其余處方量藥材,按正交表各項實驗,然后按《中國藥典》2005版I部附錄揮發油測定法提取揮發油,冷卻后稱取其量[3]。正交實驗結果見表2,方差分析結果見表3
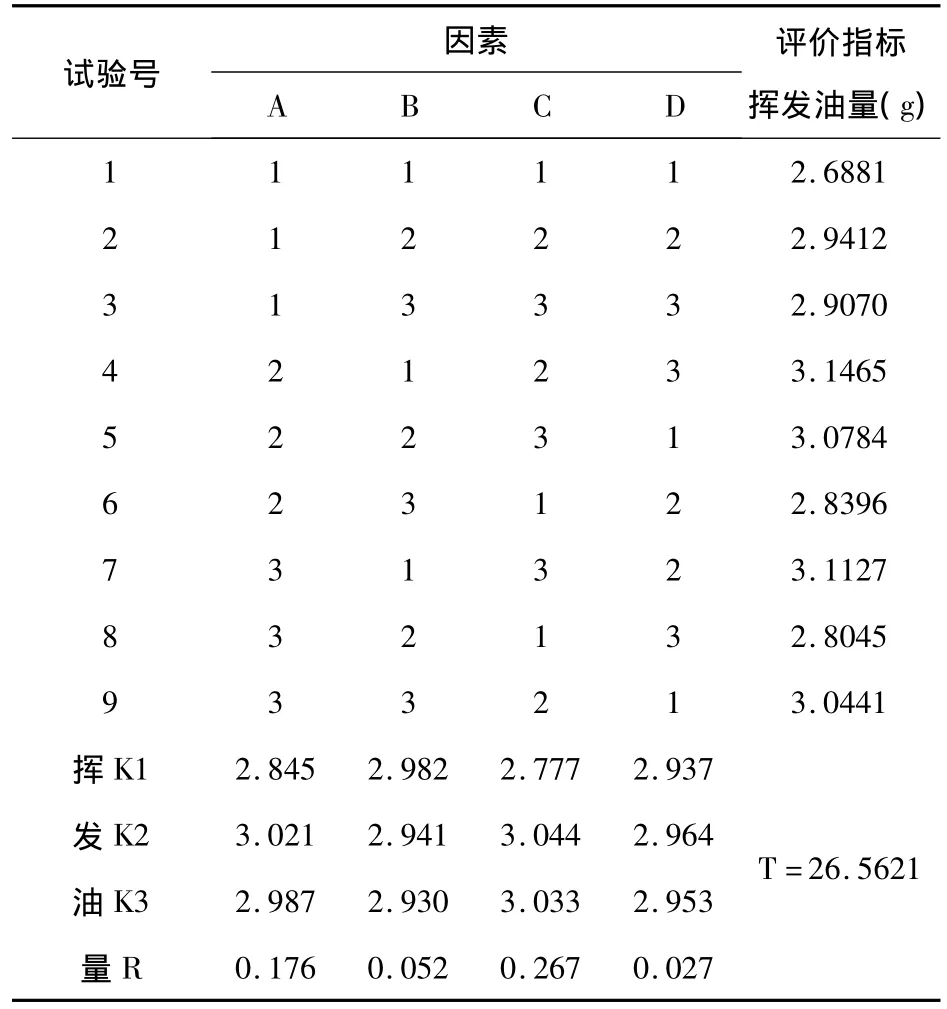
表2 正交實驗結果表
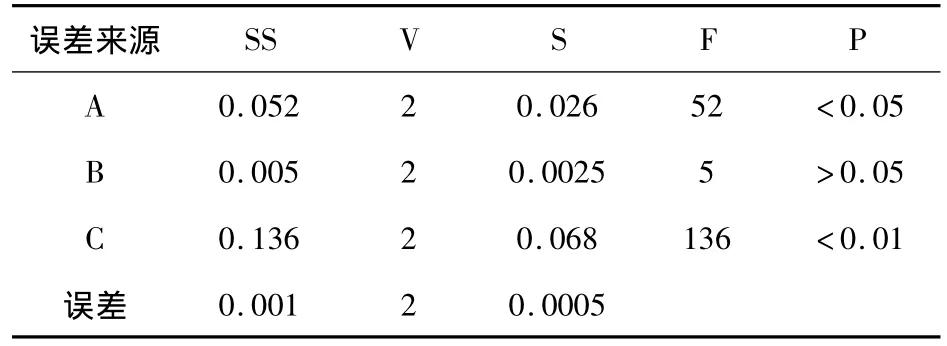
表3 正交實驗結果表
以揮發油得量為考察指標,由表2中極差R值大小顯示,各因素作用主次為C>A>B>D;表3中方差分析結果表明,C因素的影響具有及顯著性意義(P<0.01),A因素的影響具有顯著性意義(P<0.05),以A2C2為佳;B因素的影響無顯著性意義(P>0.05),因此以A2B1C2組合為佳,即藥材加10倍量的水浸泡1h,提取5h。
2.2.3 驗證試驗
按照A2B1C2最佳工藝方案驗證三批,結果見表4。

表4 乙醇回流驗證試驗結果
驗證試驗結果表明,所篩選的揮發油提取工藝穩定可行。經過不同提取比例的比較,發現最佳提取工藝為藥材加10倍量的水浸泡1h,提取5h。
2.3 揮發油包合工藝的篩選和確定:為盡可能地保留揮發油成分,保證療效,采用β-CD包合揮發油。為了適應生產,采用了操作簡便,設備要求不高的飽和水溶液法,對包合時β-CD與油的配比,包合溫度及包合時間進行正交實驗研究,以β-CD與油的配比(A)、包合溫度(B)、包合時間(C)為考察因素,設計三個水平,以揮發油利用率為考察指標,設計L9(34)正交實驗,篩選最佳包合工藝條件。
2.3.1 揮發油包合試驗因素水平表,見表5。

表5 揮發油包合試驗因素水平表
2.3.2 試驗方法及結果
按L9(34)正交表安排實驗,精密稱取 β-CD,置150mL具塞三角瓶中,加入100mL蒸餾水,水浴加熱使溶解,降至規定溫度,用磁力攪拌器攪拌30min,精密稱取揮發油1g,按1:4比例用無水乙醇稀釋,用滴管將揮發油稀釋液注入β-CD溶液中,加塞,恒溫攪拌至規定時間。置冰箱中放置24h,抽濾,用石油醚30mL洗滌三次,置干燥器中12h,即得粉末。將制得的包合物置500mL圓底燒瓶中加入蒸餾水200mL,按揮發油測定法測定包合物中實際含油量;同時進行揮發油空白回收率測定,即將1g揮發油置500ml圓底燒瓶中,按上述方法測定,計算空白回收率;在測定揮發油含量及空白回收率基礎上,可計算揮發油利用率,即:揮發油利用率(%)=包合物中實際含油量/揮發油加入量/空白回收率×100%,結果見表6。
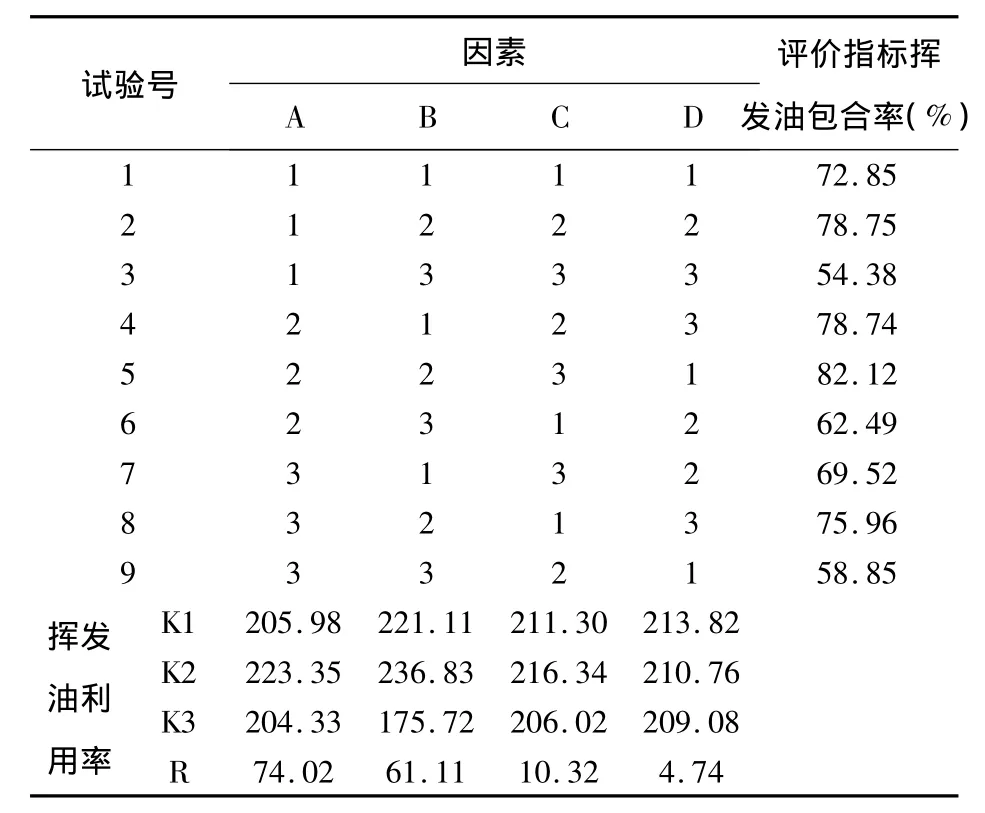
表6 正交實驗表及結果
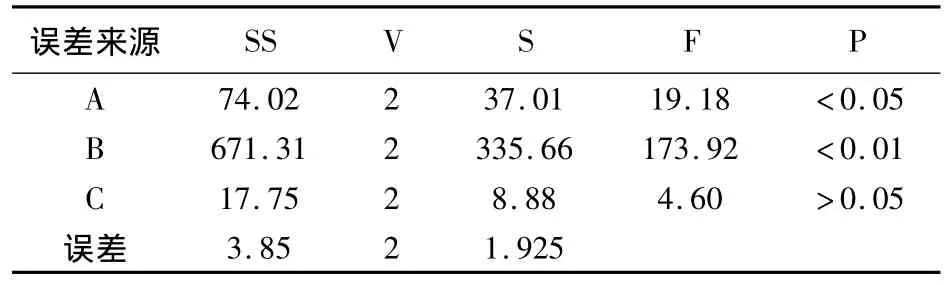
表7 揮發油β-CD包合正交實驗方差分析表
揮發油利用率越高,包合效果越好。因此,以揮發油利用率為考察指標,由表6中極差R大小顯示,各因素作用主次為B>A>C;方差結果表明:B因素的影響具有極顯著意義(P <0.01),A因素影響具有顯著性意義(P <0.05),C因素的影響無顯著意義(P >0.05),但C2稍優于C1,故以A2 B2 C2組合為佳,即以8倍揮發油量的β-CD包合,包合溫度為50℃,包合時間為3h。
2.3.3 驗證實驗
按最佳包合工藝進行揮發油的包合驗證實驗,其結果見表8。由表8中結果表明,揮發油的包合工藝穩定可行。
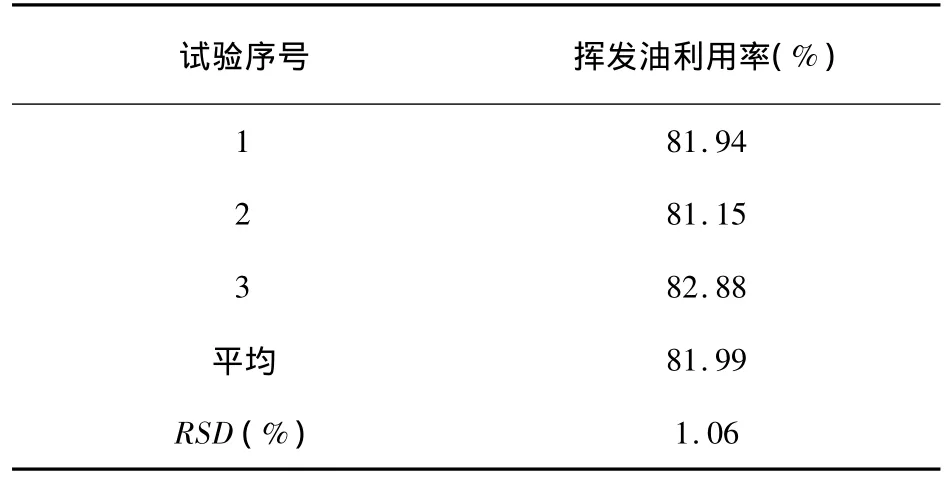
表8 揮發油包合驗證實驗結果表
2.4 揮發油包合物的定性、定量試驗
2.4.1 揮發油包合物薄層定性鑒別試驗方法及結果
取揮發油0.15g置100mL量瓶中,加石油醚定容至刻度,作為供試品溶液1;取含0.15g揮發油的β-CD包合物約1.2g,加100mL石油醚洗脫,濾液加石油醚定容至100mL,作為供試品溶液2;繼續加100mL石油醚洗脫,濾過,濾液加石油醚定容至100mL,作為供試品溶液3;取石油醚提取后的β-CD包合物加95%乙醇回流提取30min,濾過,濾液加乙醇定容至100mL,作為供試品溶液4;取β-CD1g,加95%乙醇回流提取30min,濾過,濾液加乙醇定容至100mL,作為陰性對照溶液。吸取上述5種溶液各5mL,點于同一硅膠G薄層板上,用石油醚-醋酸乙酯(17:3)為展開劑,展開,取出,涼干,置紫外光燈(365nm)下檢視,結果供試品溶液色譜中,在與揮發油溶液相應的位置上,顯相同顏色的斑點,而供試品溶液與陰性樣品色譜中無此斑點。
2.4.2 揮發油包合物紫外定性鑒別試驗方法及結果。
樣品1:精密稱取揮發油1g,置250mL容量瓶中,加無水乙醇稀釋至刻度,搖勻,備用。
樣品2:精密稱取揮發油包合物9g,用250mL石油醚分5次洗脫(50mL×5),收集洗脫液,低溫揮干石油醚,加乙醇溶液備用。
樣品3:經石油醚洗脫的揮發油包合物低溫干燥,加250mL乙醇回流提取一小時,濾過,收集濾液備用。
樣品4:精密稱取β-CD 8g,加250mL乙醇回流提取1h,濾過,收集濾液備用。
取上述樣品各10mL進行紫外掃描,結果見表9。
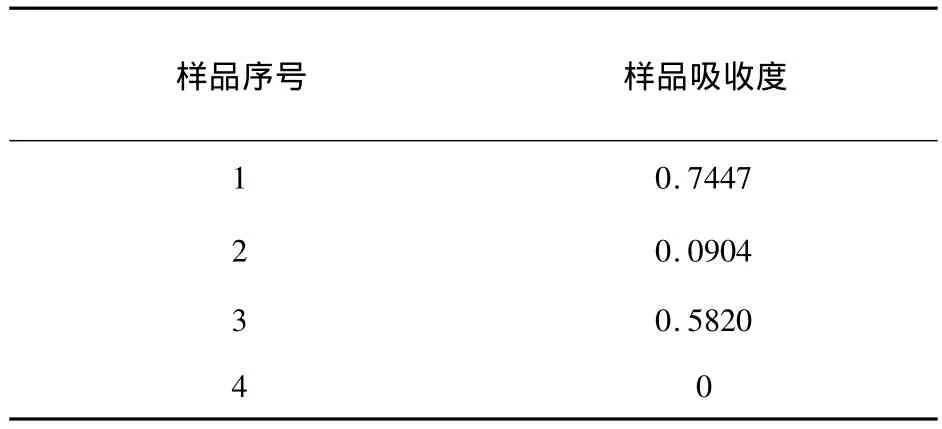
表9 揮發油包合物紫外鑒別試驗結果表
由表9中樣品吸收度結果表明:2號樣品吸收度相對于1、3號樣品比較小,說明2號樣品是由石油醚洗脫的β-CD表面殘留的揮發油,乙醇回流提取已形成包合物的揮發油,故揮發油紫外掃描圖與乙醇提取液形成的吸收光譜類似,說明包合物已形成。
綜上所述,揮發油的提取,包合工藝相對穩定可行
2.5 水提取工藝研究設計:按照工藝設計要求,對除寒水石外的其余藥材進行水提取工藝研究,以溶劑用量(A)、回流時間(B)、回流次數(C)為考察因素進行正交試驗,并以浸膏得率為評價指標,考察最佳提取工藝條件。
2.5.1 因素水平表:見表10。
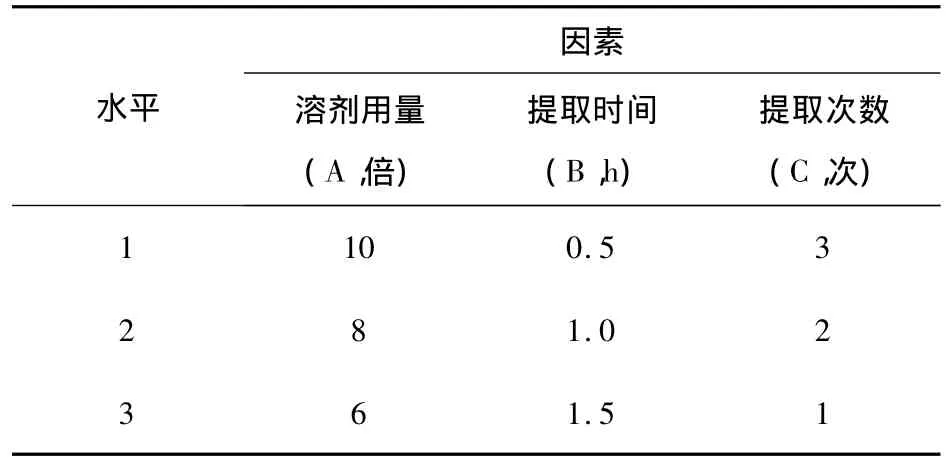
表10 水提取正交試驗因素水平表
2.5.2 實驗方法
稱取處方藥材共分9份,按因素水平表安排實驗,分別用水進行提取,提取液濃縮,蒸干,稱量計算即得。實驗設計及結果見表11
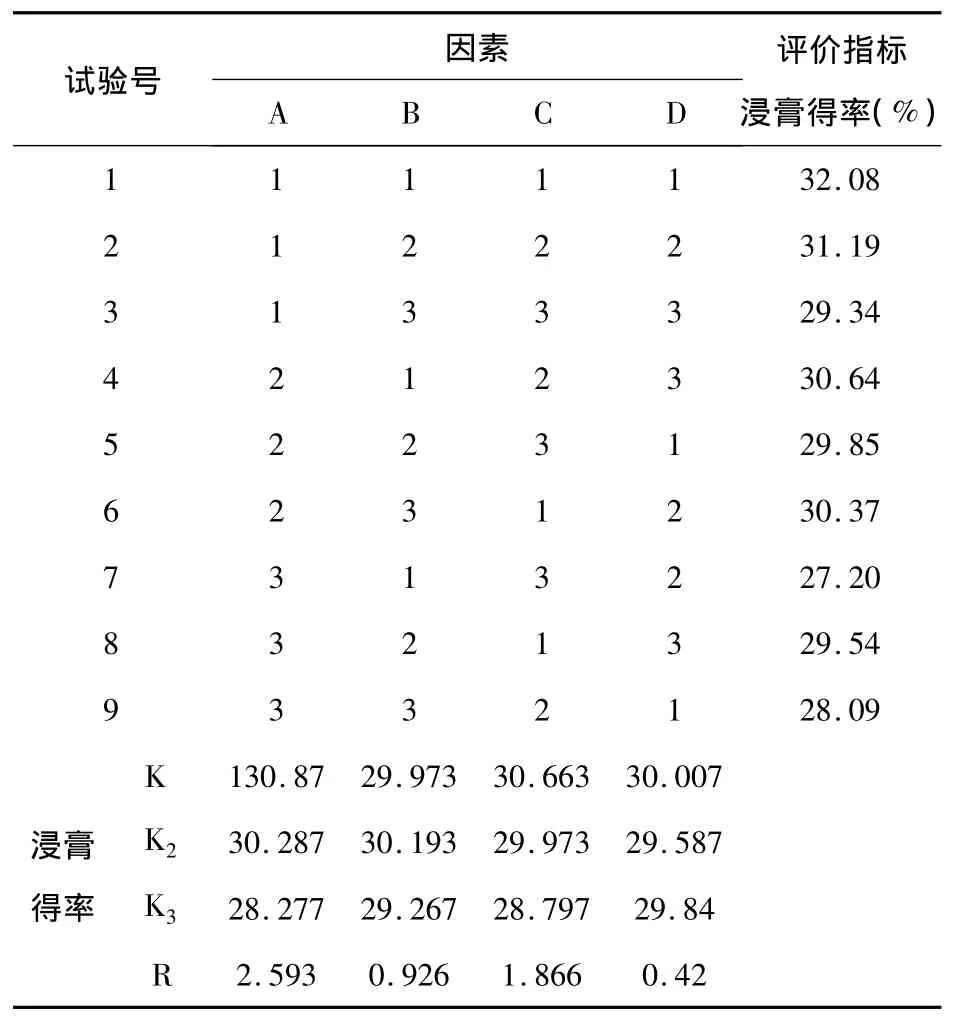
表11 正交實驗設計表及結果
2.5.3 將正交實驗結果進行方差分析,結果見表12。
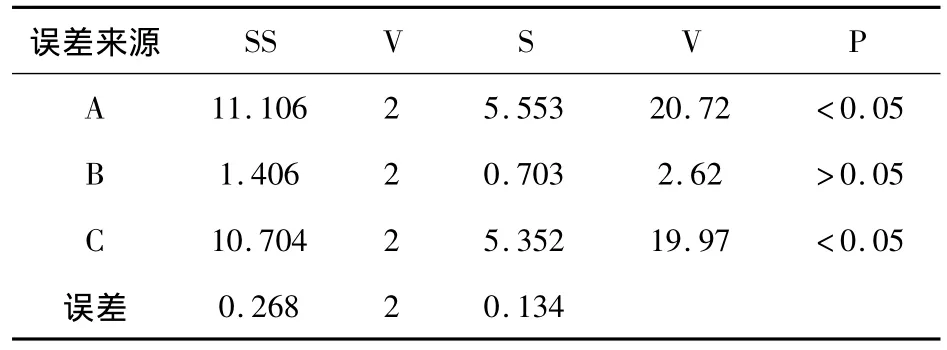
表12 浸膏得率方差分析表
由表11中極差R值大小顯示,各因素作用主次為A>C>B,方差分析結果表明,A因素和C因素的影響均具有顯著意義(P<0.05),B因素的影響無顯著意義(P>0.05),故以A1B2C1組合為佳,即以10倍量水提取1h,共提取3次。
2.5.4 提取工藝驗證試驗
稱取1/9處方量藥材按照A1B2 C1進行驗證試驗,結果見表13。結果表明:正交實驗確定的水提取工藝,穩定可行。

表13 水提工藝的驗證試驗
2.6 醇處理工藝研究設計:本品處方劑量較大,為減少服用量,保證藥物療效,應將水提膏中混有的淀粉、黏液質等雜質進行純化處理。結合大生產的生產條件,選用醇沉法,影響醇沉的主要因素有乙醇濃度、水提濃縮液的相對密度、靜置時間。以上述三個因素為考察因素,各取3個水平進行L9(34)正交試驗。并以醇浸膏得率和阿魏酸保留率為評價指標,篩選最佳醇沉工藝條件。
2.6.1 因素水平表見表14。
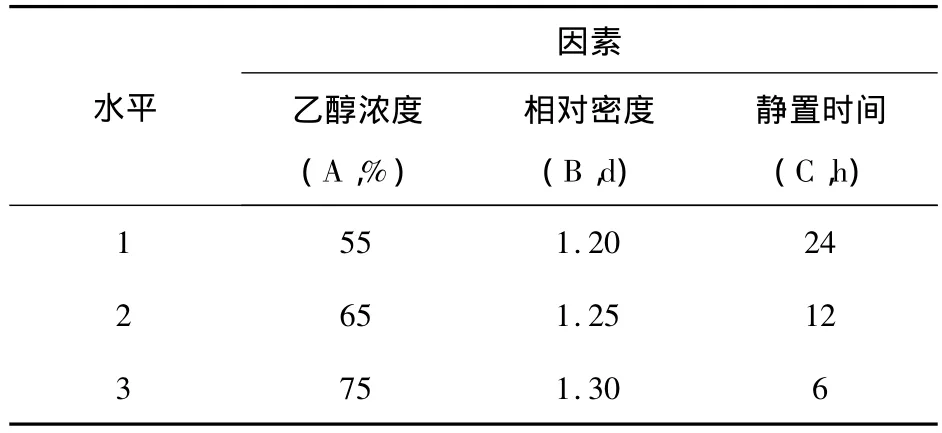
表14 醇沉正交試驗因素水平表
2.6.2 實驗方法
稱取處方量藥材分為9份,按篩選的最佳水煎工藝條件提取,將提取液濃縮成不同相對密度的濃縮液,按正交試驗條件進行醇沉、過濾,回收乙醇,減壓干燥。①浸膏得率測定:精密稱定干燥物,計算,即得。②阿魏酸保留率測定:精密稱取干燥物約0.5g,依“阿魏酸方法”進行含量測定,并計算阿魏酸保留率,即得。③正交實驗設計及結果見表15。
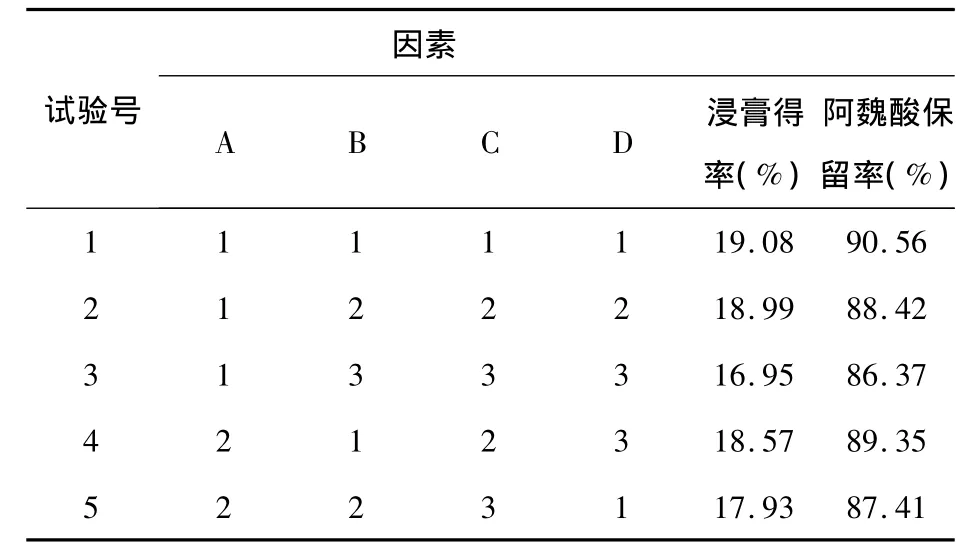
表15 醇沉正交實驗設計及結果
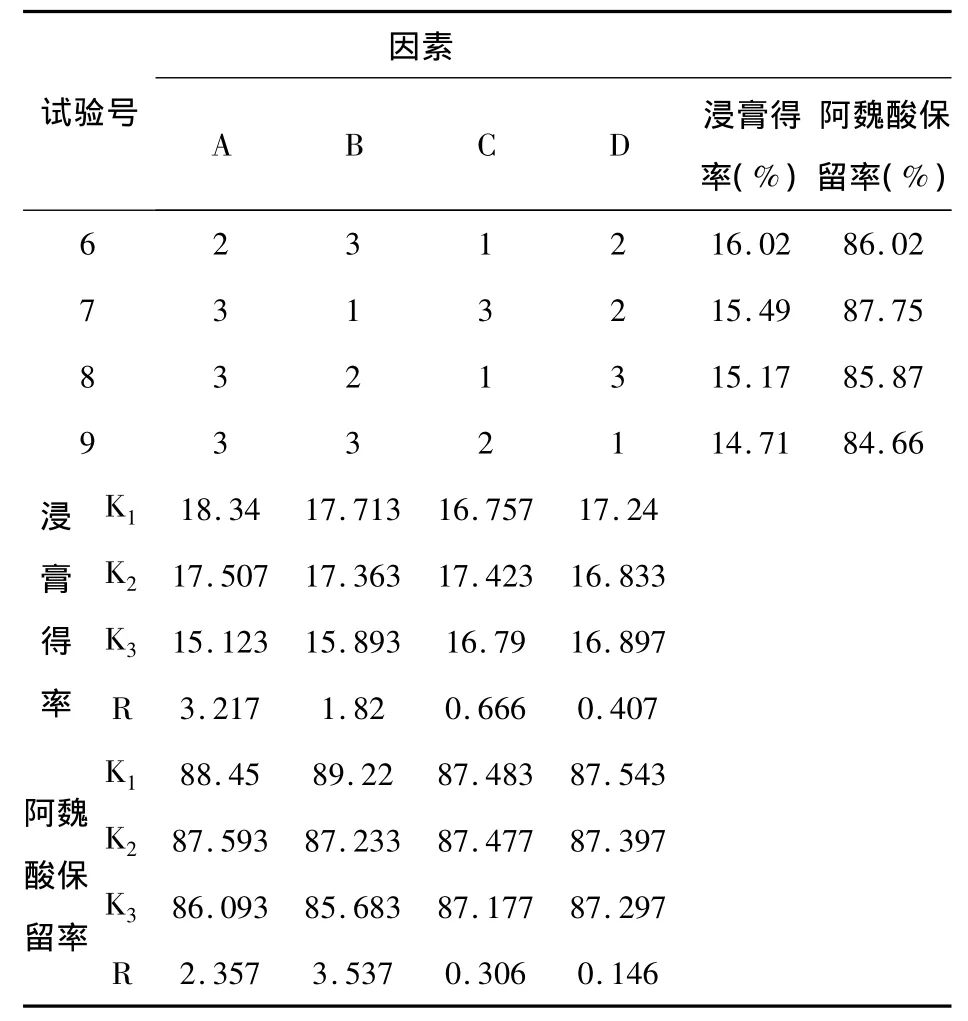
表15 (續)
④將正交實驗結果進行方差分析,結果見表16,17。
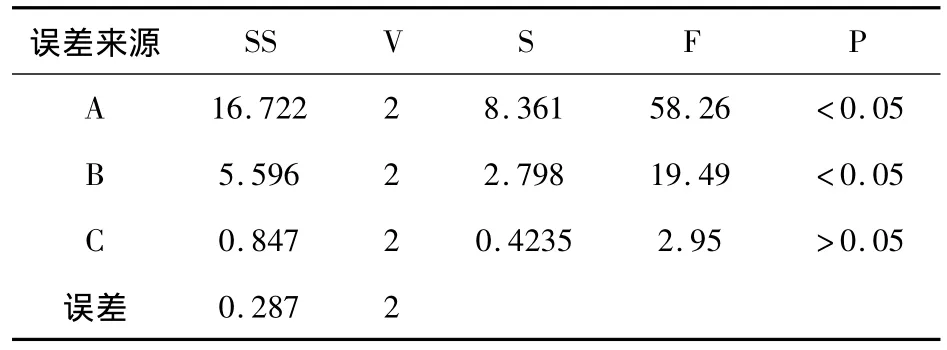
表16 醇沉法浸膏得率方差分析表

表17 阿魏酸保留率方差分析表
以醇沉后浸膏得率為考察指標,由表15中極差R值大小顯示,各因素作用主次為A >B>C;方差分析結果表明:A因素和B因素的影響均具有顯著性意義(P<0.05),C因素的影響無顯著性意義(P>0.05),考慮到應以醇沉后浸膏率少為宜,故以A3B3 C2組合為佳;以阿魏酸保留率為考察指標,由表15中極差R值大小顯示,各因素作用主次為B>A>C;方差分析結果表明:A因素和B因素的影響均具有顯著性意義(P<0.05),C因素的影響無顯著性意義(P>0.05),故以A2B3 C1組合為佳;考慮到阿魏酸保留率指標的重要性,故以A2B3 C1為最佳醇處理工藝。即藥液濃縮至相對密度為1.30,使藥液含65%乙醇,靜置24h。
2.6.3 驗證試驗
按照A2B3C1最佳工藝方案驗證三批,結果見表18。驗證試驗結果表明,所篩選的提取工藝穩定可行。

表18 醇處理驗證試驗結果
2.7 成型工藝研究
2.7.1 實驗前準備:將醇提膏干燥,粉碎,過120篩,與過200目篩的寒水石及揮發油包合物混和,備用。一個處方混合物大約為318g。
2.7.2 藥物與基質配比的初選實驗設計:為了盡量減少基質的用量,以減小服用量,同時也為后續研究工作提供依據,根據藥物性質選擇基質分別為PEG4000和PEG6000,藥物與基質的配比分別為 1:1.5,1:2.0,1:2.5,1:3.0,1:3.5,1:4.0進行滴制試驗。以滴制難易程度和滴丸圓整度為依據進行篩選,實驗結果見表19。根據實驗結果可知,藥物與基質的比例最佳為1:2.5。
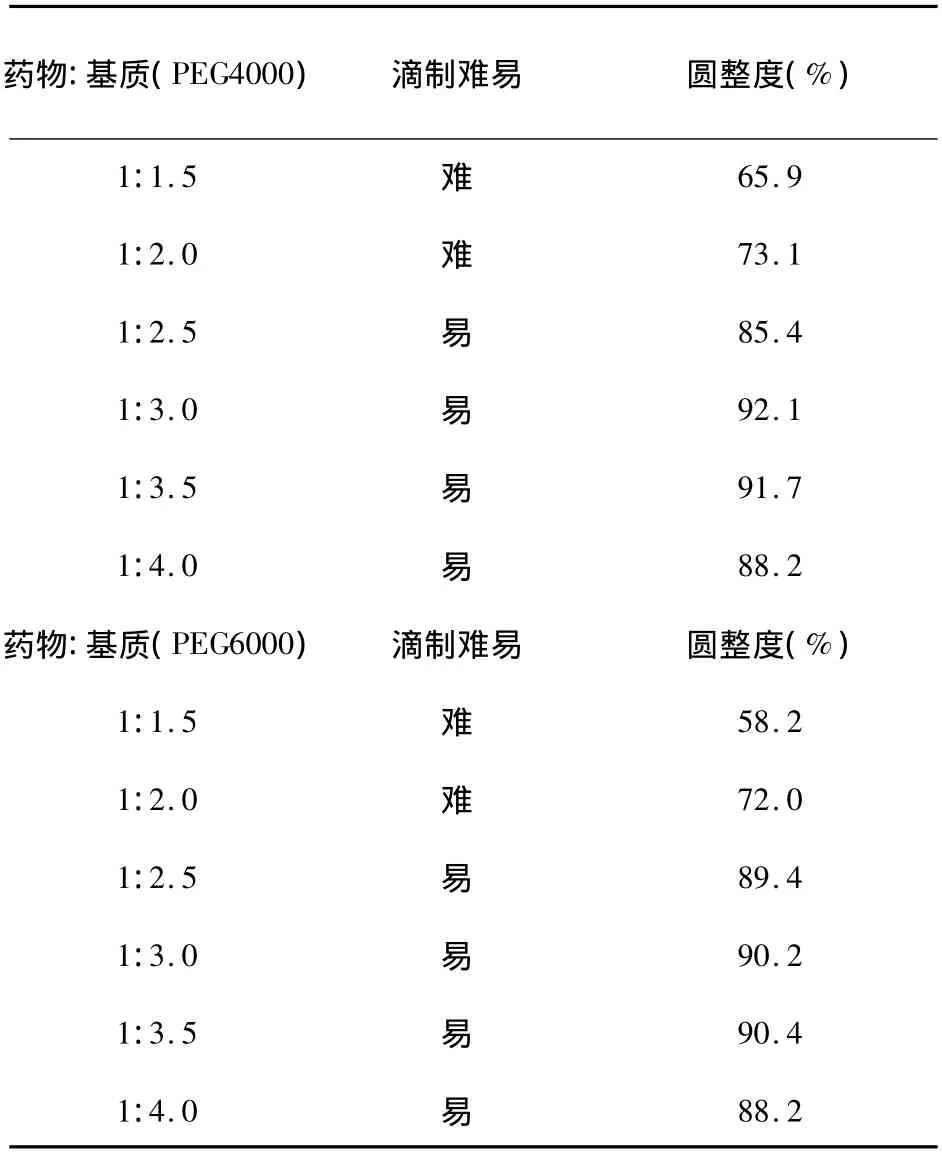
表19 藥物與基質配比的初選
2.7.3 復合基質配比的選擇
按藥物與基質配比為 1:2.5,考察 PEG4000與PEG6000 按2:0.5,2:1,2:2,2:3,2:4,2:5 的配比進行滴制試驗。以藥液黏度、滴丸圓整度與溶散時限為依據進行篩選,實驗結果見表20。根據實驗結果可知,復合基質比例最佳為2:1。
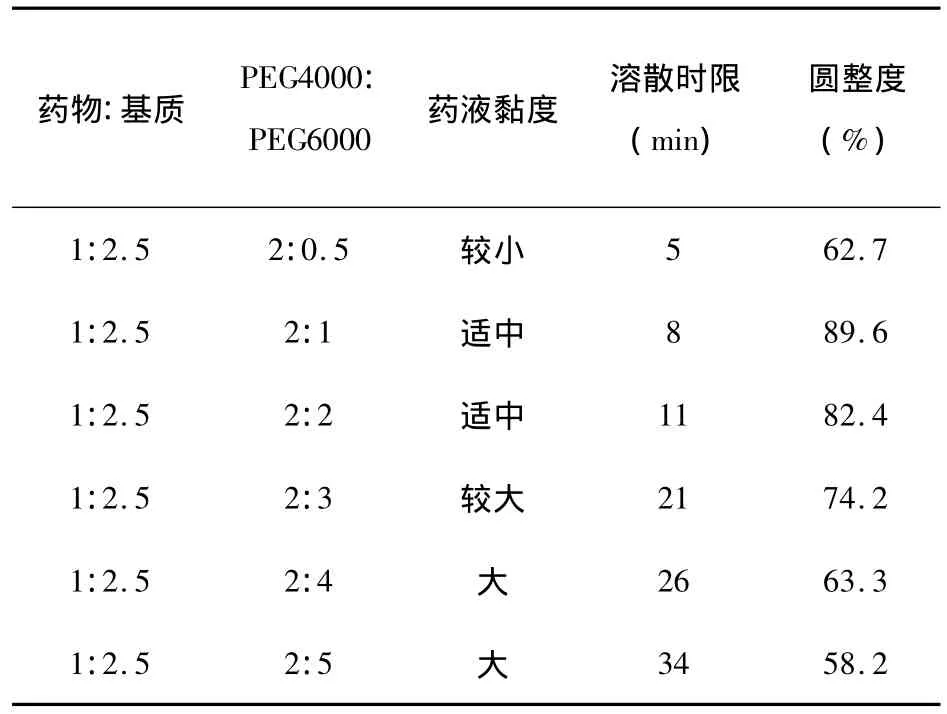
表20 復合基質配比的選擇
2.7.4 冷卻劑的選擇:根據確定的藥物與基質的配比,進行預試驗,以液體石蠟為冷卻劑能滿足滴丸劑的需求,因此確定滴丸冷卻劑為液體石蠟。
2.7.5 滴制工藝實驗研究:在單因素篩選的基礎上,以基質種類、藥物與基質配比、藥液溫度、冷卻劑溫度作為考察因素,各取3個水平進行L9(34)正交試驗。并以丸重變異系數,溶散時限和外觀質量為評價指標,篩選最佳滴制工藝條件,因素水平見表21。
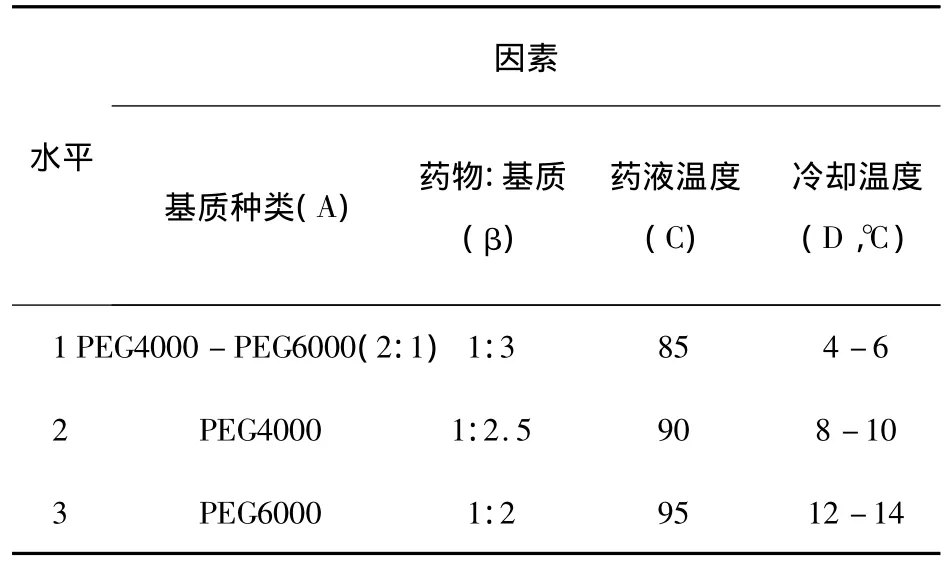
表21 滴制工藝篩選因素水平表
①正交實驗方法及結果:按照L9(34)正交表安排實驗。將基質置水浴中加熱攪拌至全部熔融后,加入規定比例的藥物,攪勻,迅速移至已預熱至恒溫的滴丸機滴料罐中,保持規定溫度,將藥液滴入液體石蠟冷卻劑中。取出滴丸,吸除表面液體石蠟,于干燥器中放置24 h,即得實驗樣品。每次取實驗樣品按《中國藥典》(2005年版)方法測定溶散時限,然后隨機抽取20粒,精密稱定總重量,再分別精密稱定各丸的重量,求出平均丸重的變異系數,并用10分制對包括滴丸成型性、外形、圓整度、硬度和色澤均勻度在內的外觀質量評分。結果見表22
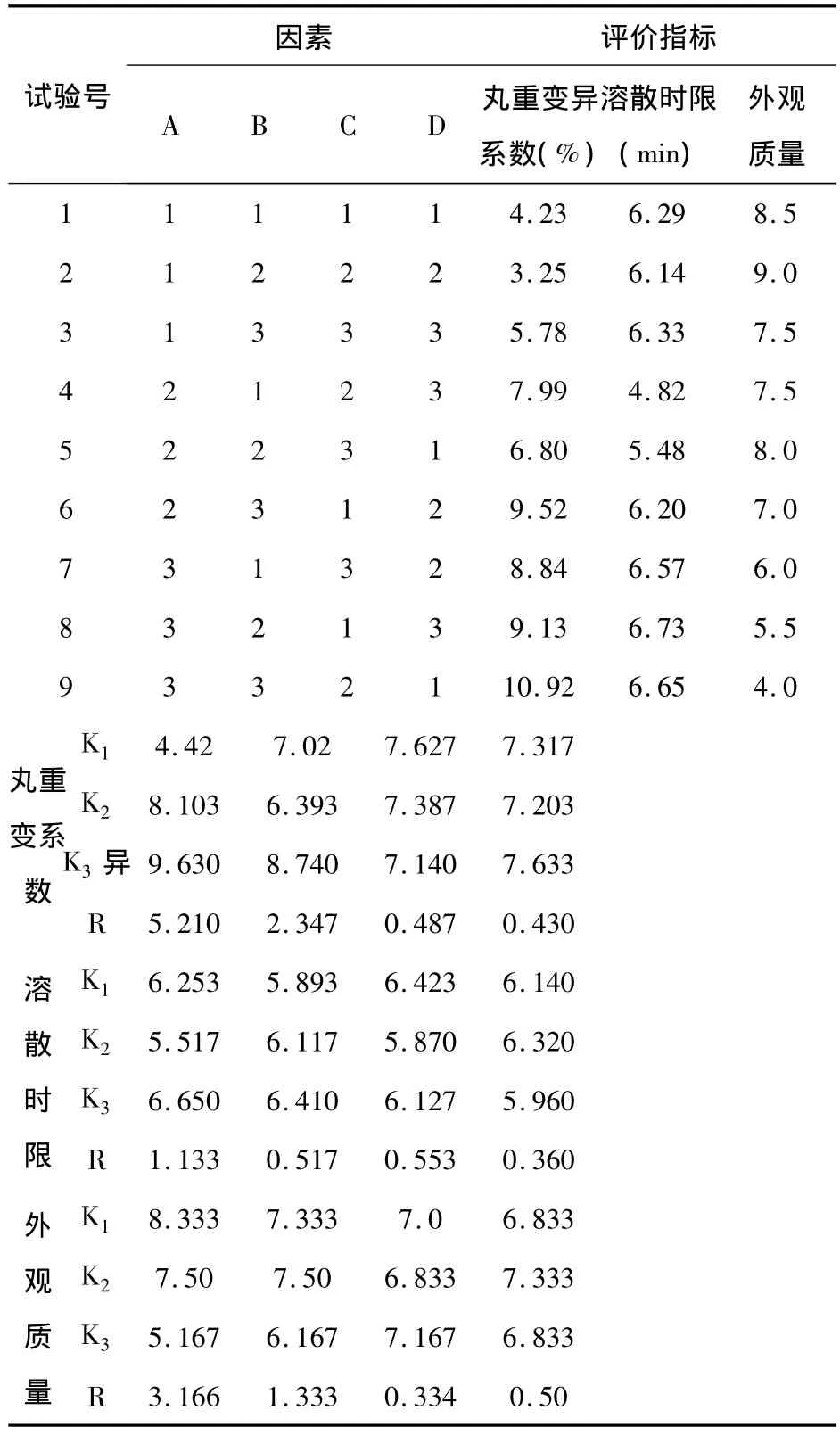
表22 滴制工藝正交實驗安排及實驗結果
②實驗結果分析:滴丸的質量不能僅用一個指標衡量,本實驗采用3個評價指標進行數據處理,求得不同指標下各因素的極差(R),從R值大小判定影響因素的主次。若選用各因素不同水平下,滴丸丸重變異系數最小,溶散時限最短,外觀質量分最高為最佳搭配 ,則3個指標下可供選擇的最佳工藝條件,分別歸納于表23中。

表23 正交實驗分析結果表
綜合表23中各指標下最佳工藝搭配,選擇兩個或兩個以上指標中均為較佳的工藝條件,得優選工藝條件為A1B2C3D2,即以PEG4000-PEG6000(2:1)為基質,藥物與基質配比為 1:2.5,藥液溫度為95℃,冷卻劑溫度為 8~10℃。
③驗證實驗:以上述確定的最佳工藝條件重復制備3批滴丸樣品,并按《中國藥典》2005年版制劑通則滴丸劑項下進行質量檢查。結果見表24。
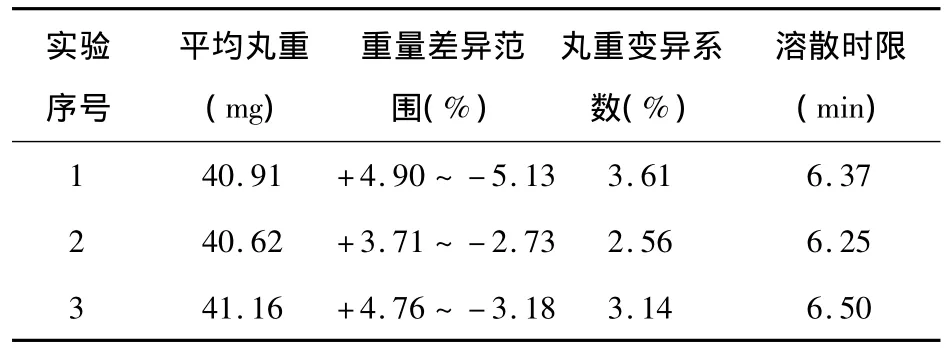
表24 滴丸滴制工藝驗證實驗結果表
驗證結果表明:驗證結果與正交實驗結果相近,說明滴丸制備工藝基本穩定可行。
3 愈心安神滴丸制備工藝總結
六味藥中,除寒水石外,其余藥材提取揮發油,用β-環糊精包合,備用。藥渣加水煎煮3次,濾過,合并濾液,濾液濃縮成相對密度為1.26~1.28(50℃)的清膏,加乙醇使含醇量達65%,攪勻,靜置24h,取上清液,回收乙醇,濃縮干燥,粉碎并過120目篩,備用,寒水石研細,過200目篩。取聚乙二醇4000與聚乙二醇6000適量加熱使溶解,加入上述揮發油包合物,稠膏(干)和寒水石研細,混勻,滴入冷卻的液體石蠟中,制成滴丸,即得。
[1]謝秀瓊.中藥新制劑開發與應用[M].人民衛生出版社,1994.
[2]楊基森,等.中藥制劑設計學[M].貴州科技出版社,1992.
[3]中華人民共和國國家藥典委員會.中國藥典一部[M].北京:化學工業出版社,2005.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
山東冶金(2019年6期)2020-01-06 07:45:54
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
世界農藥(2019年2期)2019-07-13 05:55:12
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
石油化工應用(2014年8期)2014-03-11 17:40:03
