4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′1″]三聯苯的合成
2014-02-02 08:45:57高嬡嬡趙群星閆曉亮
液晶與顯示 2014年4期
高嬡嬡,趙群星,陳 龍,李 濤,閆曉亮
(西安彩晶光電科技股份有限公司,陜西 西安 710065)
1 引 言
隨著液晶顯示技術的不斷發展,液晶顯示面板的世界市場在進一步擴大,已經廣泛地應用在電子顯示產品上,如電視、計算機屏幕、筆記型計算機、移動電話或個人數字助理等。大屏幕顯示是目前顯示市場上的普遍需求,隨著大屏幕顯示技術的發展,對彩色液晶材料也提出了更高的要求。VA-TFT模式和IPS模式由于其高對比度、寬視角以及快速響應等特點,是最具發展前景的LCD技術。在液晶材料發展趨勢中,針對LCD技術的變化,近幾年焦點都放在響應速度的提高上:要提高響應速度,液晶的黏度需要下降,或者通過液晶盒變薄來作改善,但卻會使其色彩鮮艷度受到影響。為了更好地改善液晶電視在動畫顯示方面的質量,除了通過改善顯示技術外,開發新型低粘度的液晶材料也是一條捷徑。
端烯類液晶單體與同結構烷基末端單體相比,具有黏度小、熔點低、清亮點高、低溫穩定好的優點,因而由它們調制的混合液晶具有黏度低、黏度隨溫度變化率低和低溫穩定性好的特點,目前大量使用在TFT-LCD混晶中[1-3]。丁烯類液晶是新型端烯類液晶的發展新方向,丁烯類的新型低黏度的液晶材料是改善液晶電視活動畫面顯示質量的重要途徑。目前,丁烯類液晶是高檔STN液晶和TFT混晶中一個重要組份,能顯著提高混晶的折射率各向差異性。目前丁烯類液晶的合成開發報道雖然不多[4-9],但是該類產品是TFT-LCD高檔混晶中的一個主要成分,對改進混晶的性能有著顯著作用,是一個具有廣闊前景的液晶單體。
本文擬合成化合物:4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′1″]三聯苯,是丁烯端烯類液晶中的一個代表化合物。
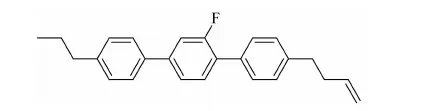
與目前液晶配方中常用的乙烯端烯類液晶相比,該化合物的分子構型的線性增加,有效改變了單體分子的長寬比,使液晶相區進一步拓寬,黏度降低、熔點降低、清亮點提高、低溫穩定性更好,并且還在三聯苯上引入側向氟原子,氟原子較高的電負性將影響到分子的偶極矩,使液晶分子具有低黏度,適中的介電各向異性、高電阻率和高電荷保持率等特點。
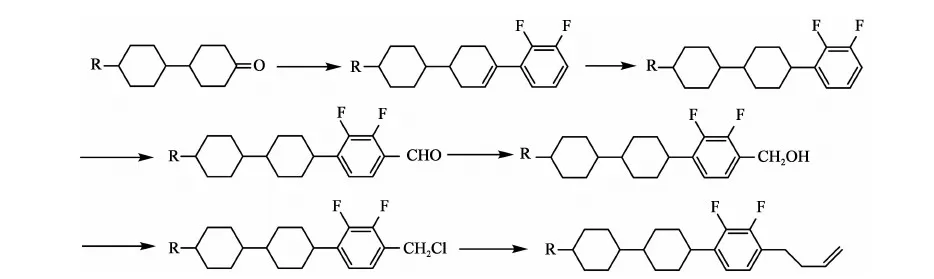
文獻報道合成丁烯端基的方法主要有2種:
方法一[5]:
方法二[10]:
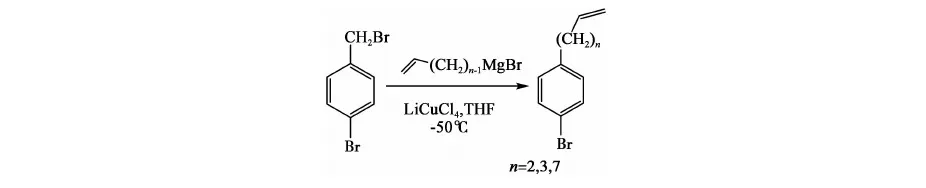
參考上述文獻及以往合成經驗,本文設計合成路線如下:
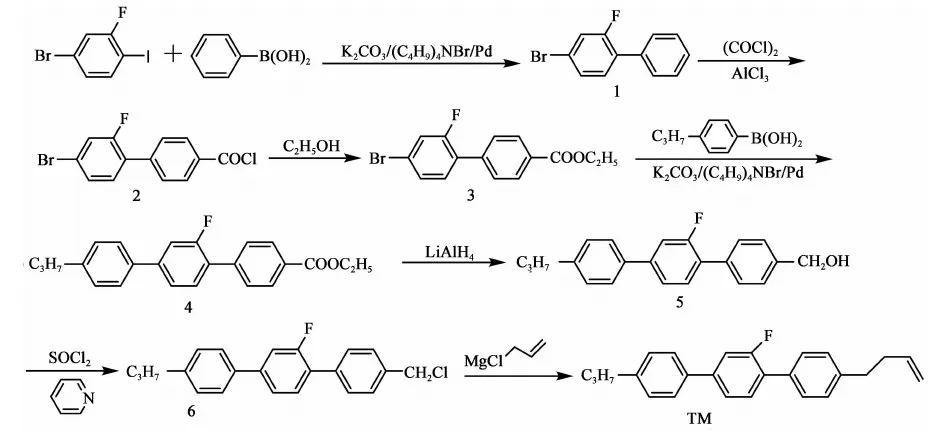
此法以2-氟-4-溴碘苯和苯硼酸為原料,經過對接、酰化、酯化反應得到2-氟-4-溴-1-乙酸乙酯基聯苯(化合物3),再與丙基苯硼酸對接、還原、氯代得到4-氯甲基-2′-氟-4″丙基-[1,1′,4′1″]三聯苯(化合物6),再與3-氯丙烯的格氏試劑對接,共七步反應得到目標產物,總收率(以2-氟-4-溴碘苯計)21.7%。
2 實 驗
2.1 原材料
原材料規格要求見表1。
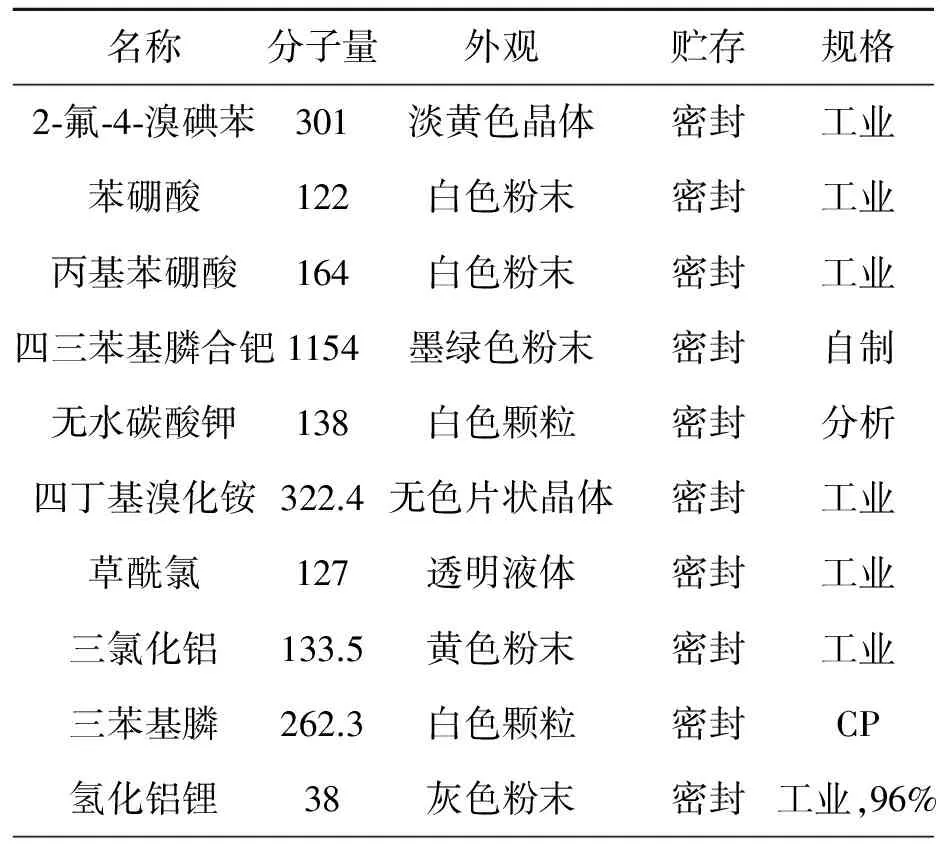
表1 原材料常數表
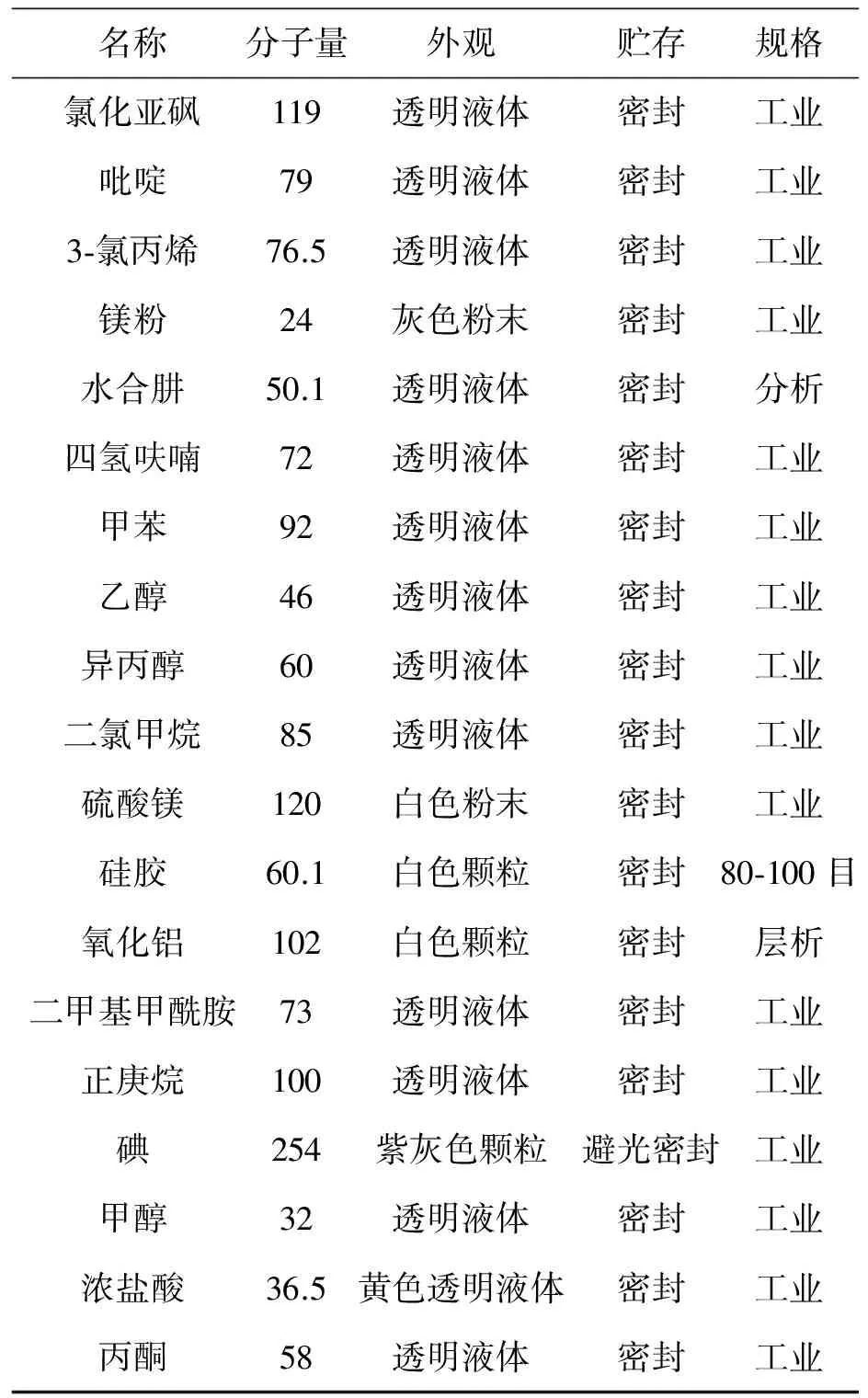
續表
2.2 儀器與設備
儀器與設備見表2。
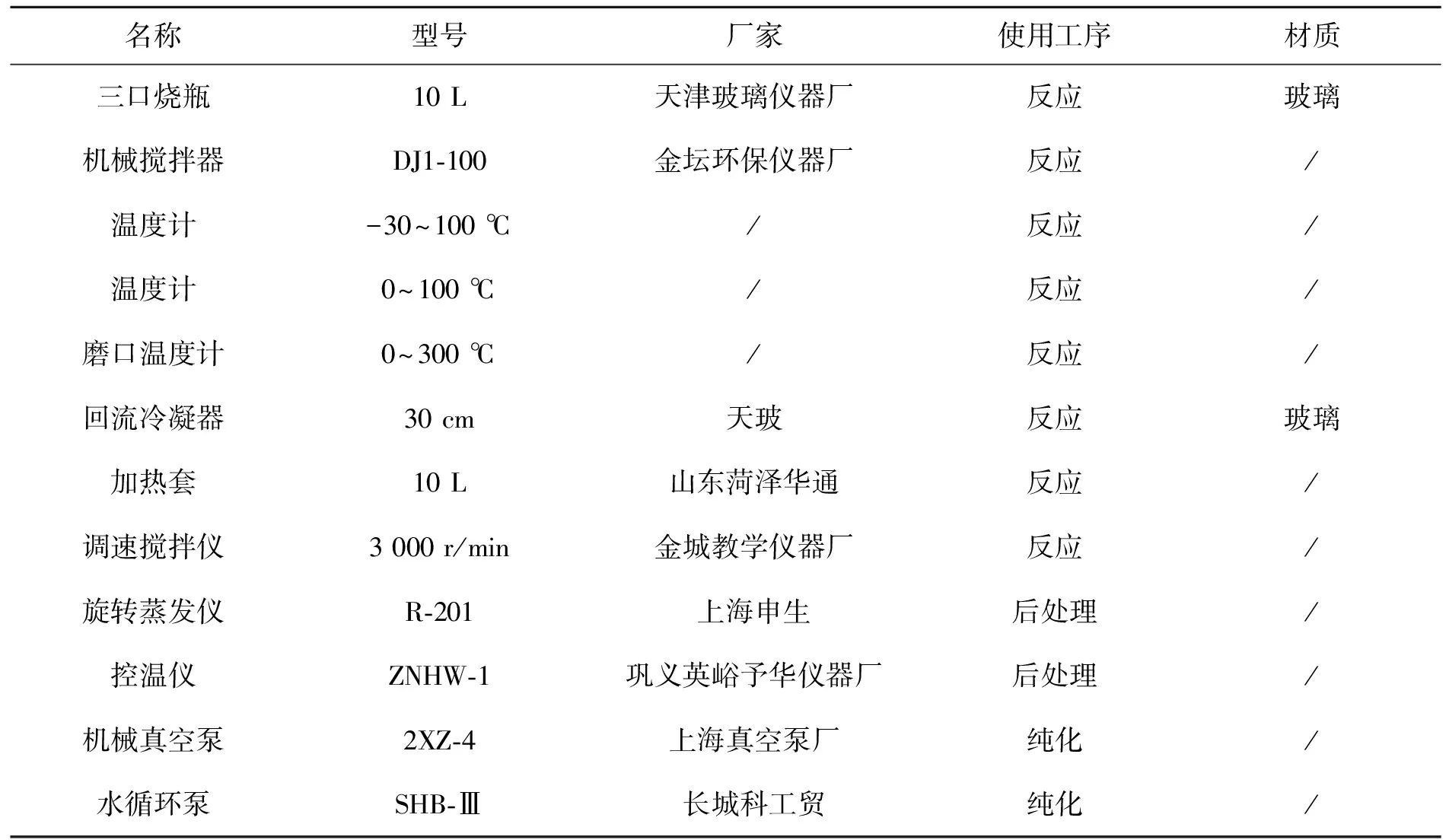
表2 儀器與設備
2.3 實驗
2.3.1 化合物1 的合成
反應方程式:
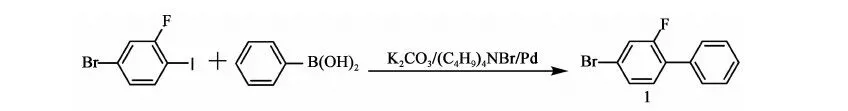
在氮氣保護下,攪拌下向10 L三口燒瓶中加入2.1 kg甲苯、2.4 kg乙醇、800 g(2.66 mol)2-氟-4-溴碘苯、214.2 g(0.66 mL)四丁基溴化銨、356.7 g(2.9 mol)苯硼酸、12.27 g(0.011 mol)自制零價鈀[Pd(Pph3)4]、0.4 kg水、777.6 g(5.63 mol)碳酸鉀,加料畢,加熱至回流(70~75 ℃)反應12 h,2-氟-4-溴碘苯GC<0.5%,停止反應。降至室溫,將反應液倒入盛有7 kg水和1.9 kg甲苯的容器中,攪拌10 min,靜置,分液,保留有機相,水相用1.74 kg甲苯提取一次,合并有機相,用8 kg×3的水水洗至中性。有機相用200 g無水硫酸鎂干燥,過濾,濾餅用0.22 kg×2甲苯淋洗,濾液合并,濃縮(真空度>0.085 MPa,70~80 ℃),得褐色液體648.5 g,粗品收率97%。高真空蒸餾,收集110~112 ℃/50 Pa下餾分,得466 g油狀物化合物1:2-氟-4-溴聯苯,GC含量99.49%,收率70%(以2-氟-4-溴碘苯計)。
2.3.2 化合物2的合成
反應方程式:
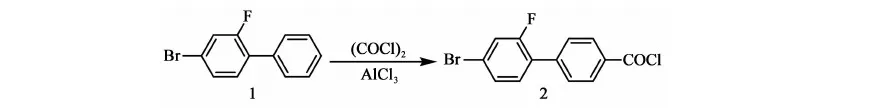
在干燥的帶有尾氣吸收裝置的10 L三口瓶中加入8.6 kg二氯甲烷,攪拌下加入860 g(6.47 mol)三氯化鋁,降溫至0~5 ℃,加入650 g(2.59 mol)化合物1,攪拌15 min,滴加658 g(5.18 mol)草酰氯,約30 min滴完,整個滴加過程溫度控制在0~5 ℃。滴畢,升溫至10~15 ℃反應1 h后取樣分析,當化合物GC含量<0.5%時,停止反應,將反應液緩慢倒入裝有2.66 kg二氯甲烷,0 ℃左右稀鹽酸(VHCl:VH2O=1∶3)的容器中,攪拌15 min,靜置,分液,有機相待用,水相用二氯甲烷提取一次,合并有機相,以8 kg×3的水,水洗至中性。有機相以300 g無水硫酸鎂干燥,過濾,濾餅用少量二氯甲烷淋洗,合并有機相,濃縮(真空度>0.08 MPa,水溫70~80 ℃),烘料(0.095 MPa,45 ℃,4 h),得黃色固體化合物2:2-氟-4-溴-1-乙酰氯基聯苯 769 g,GC含量94.3%,mp.104~112 ℃),收率94.7%(以化合物1計)。
2.3.3 化合物3的合成
反應方程式:

在干燥的,帶有尾氣吸收裝置的10 L三口瓶中加入5.2 kg甲苯,1 200 g(3.83 mol)化合物2,攪拌10 min(體系橘紅色渾濁),加入522 g(11.35 mol)乙醇,升溫至回流(78~80 ℃)反應,當化合物2 GC含量<0.5%時,停止反應。降溫至50 ℃以下,將反應液緩慢倒入裝有0.87 kg甲苯和2.4 kg水的容器中,攪拌10 min,靜置15 min,分液,有機相待用,水相用1.2 kg甲苯提取一次,攪拌10 min,靜置,分液,合并有機相,水洗至中性。有機相用200 g無水硫酸鎂干燥,過濾,濾餅用少量甲苯淋洗,合并有機相,濃縮(真空度>0.085 MPa,水溫70~80 ℃),得紅褐色液體化合物3:2-氟-4-溴-1-乙酸乙酯基聯苯1 224 g,GC含量96.1%,單步收率99%(以化合物2計)。
2.3.4 化合物4的合成
反應方程式:
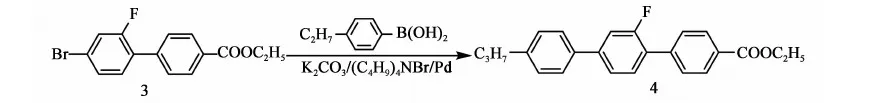
在氮氣保護下,攪拌下向10 L三口燒瓶中依次加入2.2 kg甲苯、845 g(2.62 mol)化合物3,472 g(2.88 mol)丙基苯硼酸、2.57 kg乙醇、211 g(0.65 mol)四丁基溴化銨、12.1 g(0.01 mol)零價鈀、422.5 g水、765.5 g(5.55 mol)碳酸鉀,加料完畢,加熱至回流(70~75 ℃)反應,當原料 GC含量<0.5%時,停止反應。降溫至50 ℃以下,后處理。將反應液倒入盛有7 kg水和2.5 kg甲苯的容器中,攪拌10 min,靜置,分液,保留有機相,水相用1.7 kg甲苯提取一次,合并有機相,用水洗至中性。有機相用200 g無水硫酸鎂干燥,過濾,濾餅用少量甲苯淋洗,濾液合并濃縮(真空度>0.085 MPa,70~80 ℃),得灰褐色固體917.5 g,粗品收率96.9%(以化合物3計)。向上述產品加入0.8 kg甲苯,0.82 kg乙醇,加熱至70~80 ℃,溶解后,降至室溫后,冷凍(-20 ℃,12 h),過濾,重復上述直至主含量GC>99%,產品烘干(0.095 MPa,70~80 ℃,8 h),得產品化合物4:4-乙酸乙酯基-2′-氟-4″丙基-[1,1′,4′1″]三聯苯:707.5 g,GC含量99.6%,mp.=160~163 ℃),收率74.7%(以化合物3計)。
2.3.5 化合物5的合成
反應方程式:

向10 L三口瓶中通氮氣5 min,加入1.16 kg四氫呋喃,然后分批加入48.8 g(1.28 mol)氫化鋁鋰,加料完畢,攪拌10 min后開始滴加636 g(1.76 mol)化合物4和1.7 kg四氫呋喃的混合液,控溫55~65 ℃,約30 min滴完,滴完升溫至回流(60~65 ℃),保溫30 min后取樣分析,當原料GC含量<0.1%時,停止反應,后處理。體系降溫至35~45 ℃,向體系中滴加0.08 kg丙酮,攪拌10 min向體系中加入1.74 kg甲苯,再滴加500 ml稀鹽酸(250 mL濃鹽酸+250 mL水),待體系溫度降至30~40 ℃時,倒入20 L桶中。向體系中加入1.7 kg甲苯,7 kg水攪拌10 min,靜置,分液,有機相待用,水相用1.7 kg甲苯提取,合并有機相,用水洗至中性,有機相用250 g無水硫酸鎂干燥,過濾,濾餅用0.174 kg×2的甲苯淋洗,合并有機相濃縮(真空度>0.085 MPa,70~80 ℃),得黃色固體530 g,粗品收率94.3%。向產品中加入甲苯(1 g粗品∶3 mL甲苯),加熱至75~80 ℃,溶解后,冷凍(-20 ℃,12 h),過濾,重復上述操作直至GC>99%,烘干(0.09 MPa,40~50 ℃,8 h),得產品化合物5:4-甲醇基-2′-氟-4″丙基-[1,1′,4′1″]三聯苯:460 g,GC含量99.3%,mp.=121.5~123.4 ℃),收率81%(以化合物4計)。
2.3.6 化合物6的合成
反應方程式:
攪拌下,向干燥的1 L三口瓶中依次加入4.85 kg甲苯,930 g (2.9 mol)化合物5,277 g(3.5 mol)吡啶,加熱,待體系溫度50~55 ℃,開始滴加415 g(3.5 mol)氯化亞砜與0.36 kg甲苯的混合溶液,控溫60~70 ℃,約60 min滴完,70~75 ℃下保溫反應1 h,取樣分析,當原料LC含量<0.1%時,停止反應。降溫至50 ℃以下,靜置2 h后,分液,上層有機相在攪拌下倒入盛有1.25 kg甲苯,50 g氯化鈉,3 L稀鹽酸(1 500 mL濃鹽酸+1 500 mL水)的容器中,攪拌10 min,靜置,分液,有機相待用,水相用甲苯提取一次,合并有機相,水洗至中性,有機相用300 g無水硫酸鎂干燥,過濾,濾餅用甲苯淋洗,合并濾液濃縮(真空度>0.085 MPa,70~80 ℃),得粗品877 g,粗品收率89.1%(以化合物5計)。將上述粗品以1 g粗品∶2 mL甲苯:1 mL乙醇重結晶(-20 ℃,8 h),至LC>99.5%,得產品化合物6:4-氯甲基-2′-氟-4″丙基-[1,1′,4′1″]三聯苯:650 g,mp.=112.9~114.7),收率66.7%(以化合物5計)。
2.3.7 目標產物TM的合成
(1)氯丙烯格氏試劑的制備
反應方程式:

在氮氣保護下,攪拌下向10 L三口瓶中依次加入31 g四氫呋喃、33.4 g(1.4 mol)鎂粉,滴入少量氯丙烯使其引發(體系溫度60~70 ℃),滴加67.5 g(0.88 mol)氯丙烯,滴加過程中控制體系溫度在-10~0 ℃,約35 min滴完。滴畢在-10~0 ℃下反應0.5 h,室溫靜置1 h,將上層清液倒入1 L三口瓶中,密封待用。
(2)化合物TM的制備

氮氣保護下,將上述格氏試劑升溫至40 ℃,滴加60 g(0.18 mol)化合物6與0.24 kg四氫呋喃的混合溶液,滴加過程中溫控40~50 ℃,滴完保溫反應1 h,當原料LC含量<0.05%時,停止反應。將反應液倒入盛有230 ml稀鹽酸(80 mL濃鹽酸+150 mL水),0.26 kg甲苯的燒杯中,攪拌15 min,靜置,分液,有機相待用,水相用60 mL甲苯提取一次,合并有機相,用水洗至中性,有機相用20 g無水硫酸鎂干燥,過濾,濾餅用60 mL甲苯淋洗一次,合并濾液濃縮(真空度>0.085 MPa,70~80 ℃),得粗品63 g,粗品收率103%。將上述粗品以1 g粗品∶2 mL乙醇重結晶,過濾,濾餅用15 g乙醇淋洗,烘干(0.095 MPa,35~40 ℃,4 h),得目標產物4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′,1″]三聯苯的合成:50 g,LC含量99.9%;GC含量99.8%,收率81.9%(以化合物6計)。IR(v/cm-1):3 023,2 955,2 925,2 866,1 915,1 639(C=C),1 546,1 487,1 396,1 182,1 132,1 005,893,798;1H NMR(CDCl3,500 MHz):7.41~7.53(m,7H),7.23~7.38(m,4H),5.06~5.09(d,J=18.5 Hz,1H),4.99~5.01(d,J=10.5 Hz,1H),2.74~2.77 (t,J=7.75 Hz,2H),2.61~2.64 (t,J=7.75 Hz,2H),2.39~2.44(m,2H),1.64~1.72(m,2H),0.96~0.98(t,J=7.25 Hz,3H); MS(m/z %):344.5(M+,40),303(100),274(40)。
總收率:21.7%(以2-氟-4-溴碘苯計)。
3 結果與討論
3.1 化合物1的合成討論
本步反應重復3次,3次反應中控取樣分析結果見表3。
表3化合物1反應時間與產物含量變化表
Tab.3 Relation between content of compound 1 and reaction time

反應名稱18h產物GC含量%原料GC含量%20h產物GC含量%原料GC含量%22h產物GC含量%原料GC含量%反應一//93.91.793.40.2反應二91.71.594.60.09//反應三94.41.3091.90.04//
以上數據看出,該反應在20~22 h可終止反應。
3.2 化合物2的合成討論
本步反應重復3次,3次反應取樣分析結果見表4。
表4化合物2反應時間與產物含量變化表
Tab.4 Relation between content of compound 2 and reaction time

反應名稱1.5h產物GC含量%原料GC含量%2.5h產物GC含量%原料GC含量%反應一92.90.0197.8/反應二72.90.02920.16反應三91.00.0194.90.04
以上數據看出本反應在2.5 h可停止反應。
3.3 化合物3的合成討論
本步反應重復3次,3次反應中取樣分析結果見表5。
表5化合物3反應時間與產物含量變化表
Tab.5 Relation between content of compound 3 and reaction time

反應名稱1h產物GC含量%原料GC含量%4h產物GC含量%原料GC含量%反應一95.11.297.20.08反應二//96.00.01反應三//99.00.01
以上數據看出本反應重復性好,在4 h停止結束。
3.4 化合物4的合成討論
本步分別用鈀炭(反應一,二)和零價鈀(反應三,四)2種體系,取樣分析結果見表6。
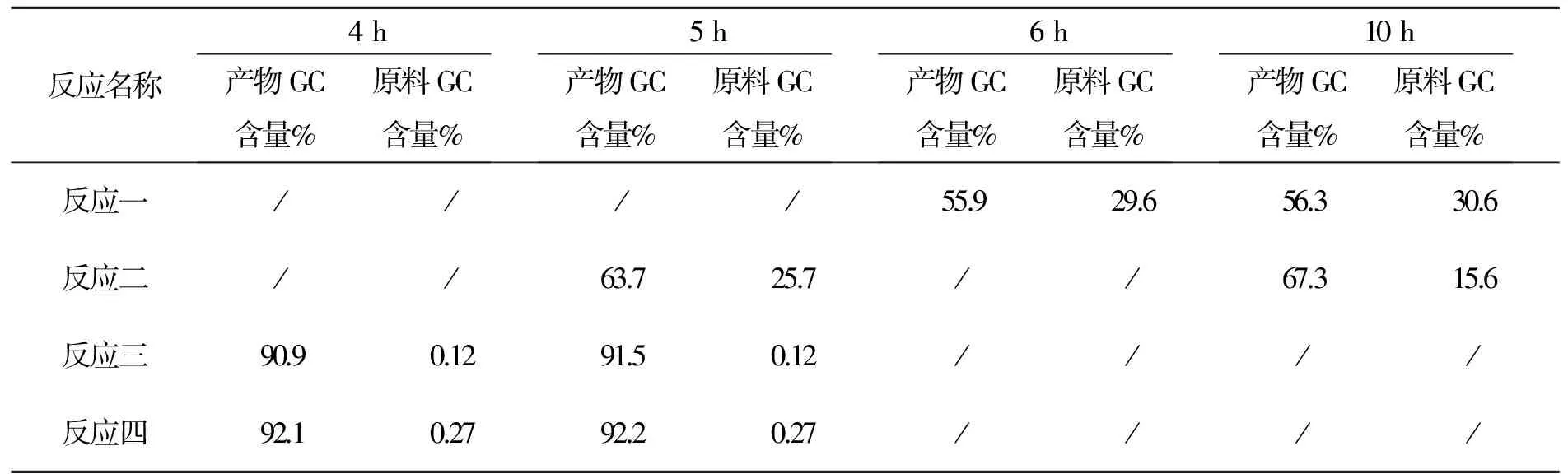
表6 化合物4反應時間與產物含量變化表
反應一:鈀炭體系∶硼酸=1∶1.1;反應二鈀炭體系∶硼酸=1∶1.8;反應三:四零價鈀體系∶硼酸=1∶1.1。
以上數據可以看出本反應零價鈀體系4 h可以反應完全,鈀炭體系硼酸比例為1∶1.8仍反應不完。
3.5 化合物5的合成討論
本步反應取樣分析結果見表7。

表7 化合物5反應時間與產物含量變化表
各組氫化鋁鋰料比,反應一1∶1.2;反應二1∶0.5;反應三1∶0.6;反應四五1∶0.7。從以上數據可以看出氫化鋁鋰比例1∶1.2時,產生掉氟較大,當氫化鋁鋰比例為1∶0.5、1∶0.6時,原料有剩余;當氫化鋁鋰比例為1∶0.7時1 h可反應完全,掉氟<0.05%。
3.6 化合物6的合成討論
本步反應取樣分析結果如下
各組實驗料比:化合物5∶SOCl2∶吡啶
反應一 1∶1∶0
反應二 1∶1∶0.3
反應三 1∶1∶1
反應四 1∶1.2∶1(4 h后補加20%氯化亞砜)
反應五 1∶3∶1.2
反應六 1∶1.2∶1.2(4 h后補加20%氯化亞砜,5 h后二次補加20%氯化亞砜)
分析結果見表8。
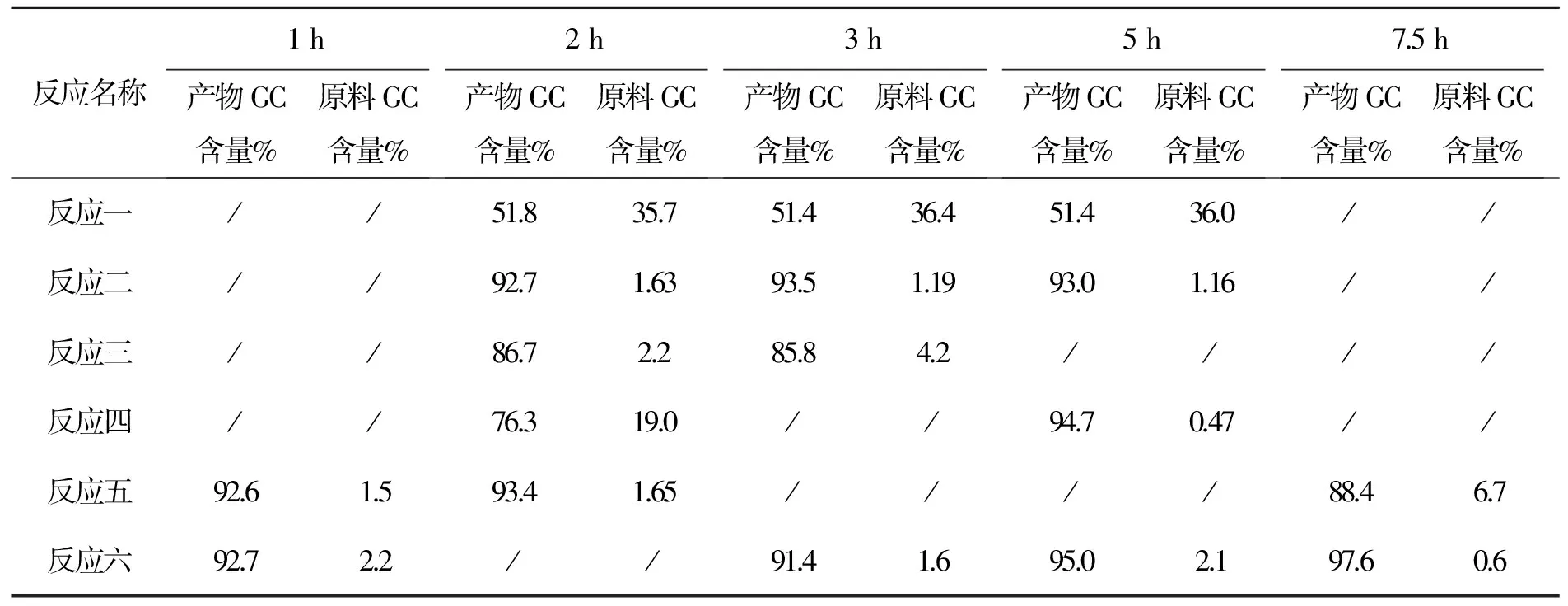
表8 化合物6反應時間與產物GC含量變化表
以上數據看出:(1)吡啶加快了反應速度。(2)當氯化亞砜調至1∶3時延長反應時間仍反應不完。(3)氯化亞砜需多次補加才能使原料反應完全,采用LC進行跟蹤,數據見表9。
表9化合物6反應時間與產物LC含量變化表
Tab.9 Relation between content of compound 6 and reaction time
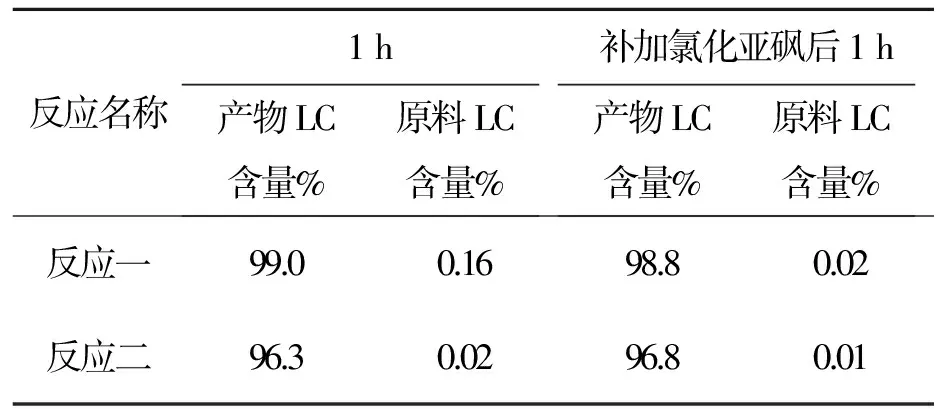
反應名稱1h產物LC含量%原料LC含量%補加氯化亞砜后1h產物LC含量%原料LC含量%反應一99.00.1698.80.02反應二96.30.0296.80.01
以上數據看出,用LC跟蹤補加氯化亞砜后原料可反應完全。
3.7 目標產物TM的合成討論
本步溫度條件反應重復兩次,兩次反應中取樣分析結果見表10。
表10化合物7反應時間與產物GC含量變化表
Tab.10 Relation between content of compound 7 and reaction time
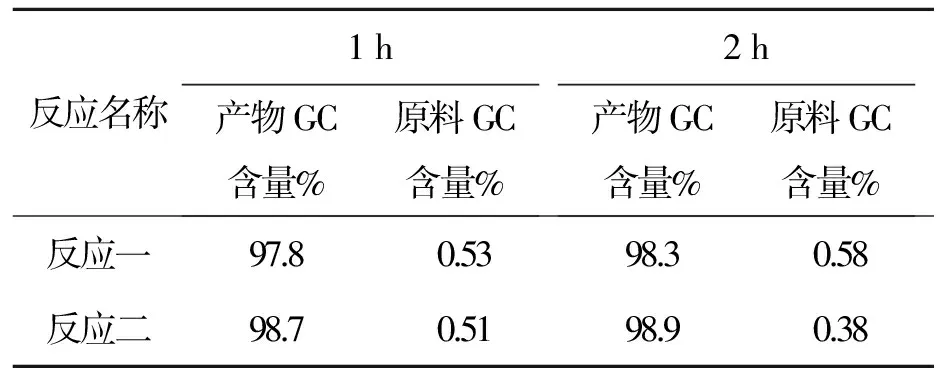
反應名稱1h產物GC含量%原料GC含量%2h產物GC含量%原料GC含量%反應一97.80.5398.30.58反應二98.70.5198.90.38
以上數據可以看出控溫35~40 ℃,原料反應至0.5%左右很難再減小。故把溫度升高,改為40~45 ℃,且用GC//LC對比,數據見表11。
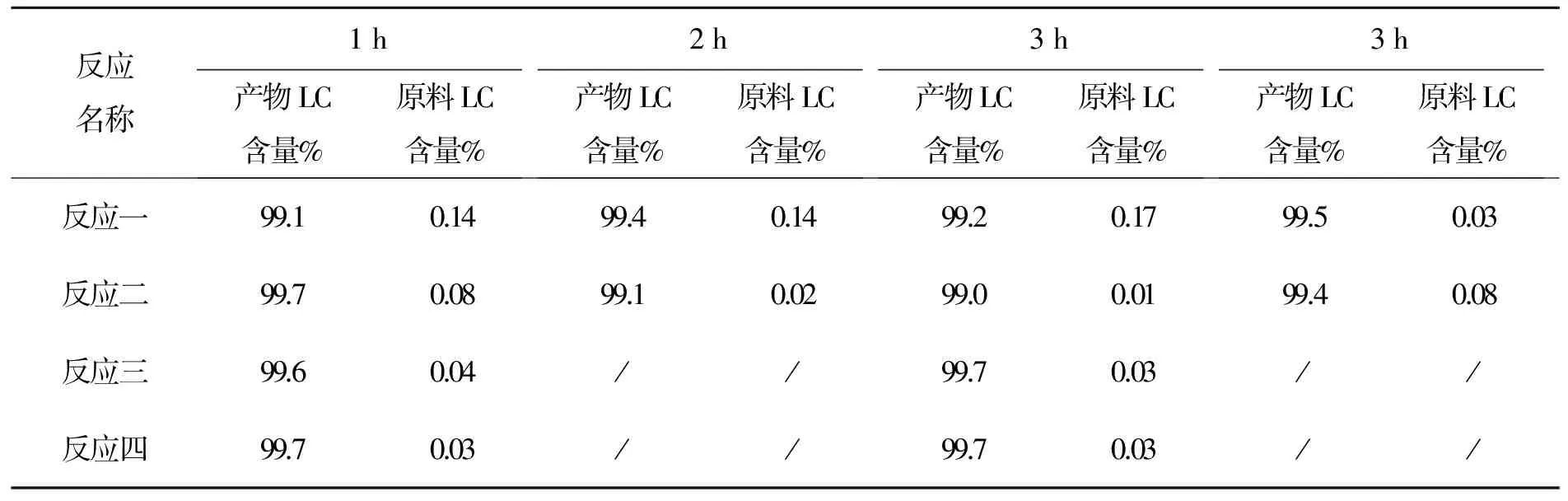
表11 化合物7反應時間與產物LC含量變化表
從以上數據看出,該反應用LC跟蹤更精確,3 h可停止反應。
4 結 論
本文以2-氟-4-溴碘苯和苯硼酸為原料,共經過7步反應得到目標產物,總收率21.7%。與底物不同的類似文獻路線相比,首次路線完整的報道了4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′,1″]三聯苯的合成過程。本文路線與文獻報道的構架丁烯端基方法一相比較,相似之處在于都進行了芐醇結構、芐氯結構的合成以及與烯丙基氯格氏試劑的反應,不同之處在于芐醇結構的來源。文獻通過鋰試劑超低溫反應水解得到苯甲醛基,然后還原制得芐醇,成本較高,且醛基易被氧化,不易保存;本文通過酰化、酯化得到苯甲酸酯基,然后還原制得芐醇,酯性能穩定,易于保存,且成本降低。與文獻報道的構架丁烯端基方法二相比較,文獻采用了芐溴基與烯丙基溴格氏試劑反應,同樣采用鋰試劑催化、超低溫反應條件(-50 ℃),本文采用烯丙基氯格氏試劑反應,溫度40 ℃左右,條件溫和,易于操作。文中詳細討論了各反應過程中反應時間對產物轉化的影響。與課題組開發的此化合物其它合成路線相比,最后一步構架丁烯端基,可較好的控制烯鍵易位,易純化。反應過程所用原料廉價易得,可有效降低成本,同時利于實現工業化生產。
[1] 員國良,鄭成武,華瑞茂. 含鏈端烯基負性液晶單體的合成及其性能研究[J].液晶與顯示,2013,28(4):510-515,551.
Yun G L,Zheng C W,Hua R M.Preparation and characteristics of terminal alkeny bearing lateral fluoro benzene negative liquid crystal [J].ChineseJournalofLiquidCrystalsandDisplays,2013,28(4):510-515,551. (in Chinese)
[2] 趙地順,李洪勝,段二紅,等. 雙烯含氟類液晶化合物的合成與表征[J].河北師范大學學報:自然科學版,2010,34(4):448-452.
Zhao D S,Li H S,Duan E H,etal.Synthesisand properties of diolefin fluorous liquid crystal compound [J].JournalofHebeiNormalUniversity:NaturalScienceEdition,2010,34(4):448-452.(in Chinese)
[3] 張婷婷,姜雪松,李永剛,等. 一種雙乙烯類液晶化合物及其制備方法:中國,CN101928199 B[P]. 2010-08 -04.
Zhang T T,Jiang X S,Li Y G,etal.Divinyl liquid crystal compound and preparation method:China,CN101928199 B[P]. 2010-08-04.(in Chinese)
[4] 胡葆華,袁鵠,周銀波,等.丁烯類化合物:中國,CN102203214 B [P].2013-07-24.
Hu B H,Yu H,Zhou Y B,etal. Butenes:China,CN102203214 B [P].2013-07-24.(in Chinese)
[5] 任惜寒,孟勁松,員國良,等.烷基雙環己基2、3-二氟苯丁烯類液晶化合物及其用途:中國,CN102153442 A [P].2011-08-17.
Ren X H,Men J S,Yun G L,etal.Alkyl dicyclohexyl 2,3-difluoro-butene liquid crystal compounds and their use:China,CN102153442 A [P].2011-08-17.(in Chinese)
[6] KELLY S M. Four unit linking groups 3 liquid crystals of negetive dielectric anisotropy [J].LiquidCrystals,1991,10(2):261-272.
[7] KELLY S M.Four unit linking groups 4 Liquid crystals of negetive dielectric anisotropy [J].LiquidCrystals,1991,10(2):273-287.
[8] SHIMADA. Liquid crystalline compound,Liquid crystalcomposition,Liquid crystaldisplay element,WO2008090780 A1[P].2008-07-31.
[9] 員國良,賈剛剛,梁志安,等. 含有雙3-丁烯基含氟三聯苯液晶化合物及其制備方法:中國,CN201110051298[P]. 2011-03-04.
Yun G L,Jia G G,Liang Z A,etal.Double 3-butene containing fluorinated terphenyl liquid crystal compound and preparation method thereof:China,CN201110051298 [P]. 2011-03-04.(in Chinese)
[10] Rajiv R S,Robert R,Singhaus G W. 4-dihydroxyborylphenyl analogues of 1-aminocyclobutanecarboxylic acids:potential boron neutron capture therapy agents [J].J.Org.Chem,1999,64(23):8495-8500.
