吡唑基鎂鹵化物/四氫呋喃可充鎂電池電解液
2014-02-18 12:06:32非路熱吐爾遜祖麗皮亞沙地克努麗燕娜王久林
物理化學學報 2014年9期
非路熱?吐爾遜 祖麗皮亞?沙地克 努麗燕娜 楊 軍 王久林
(上海交通大學化學化工學院,上海200240)
1 引言
人類社會的發展離不開優質能源的出現和先進能源技術的使用.隨著人類對能源需求日益增大,不可再生能源短缺迫使人們把目光轉向新型可再生的清潔能源方面.化學電源由于具有能量轉換效率高、能量密度大、污染小、可移動等特點備受人們青睞.在現有的化學電源技術中,鋰離子電池因其能量密度大、技術成熟而被廣泛應用于各個領域,但由于存在安全性較低和成本較高的問題,在大型電能儲存設備和電動汽車動力電池方面應用受到一定限制.人們開始注意到在元素周期表上與鋰處于對角線位置的鎂,根據對角線規則,二者具有較為相似的物理和化學性質,且鎂儲量十分豐富,具有成本低、電極電位較低、理論容量較高、加工處理方便、對環境無污染等諸多優點,因此以金屬鎂為負極的可充鎂電池被認為在大型電能儲存設備和電動汽車動力電池方面很有發展前景.1,2
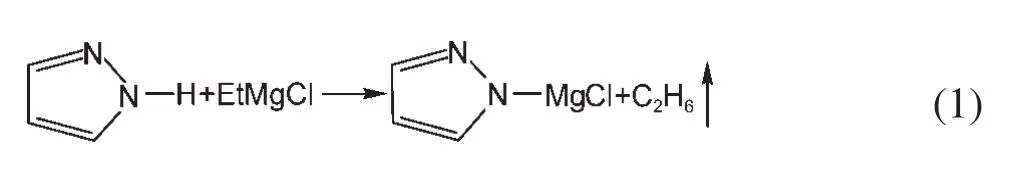
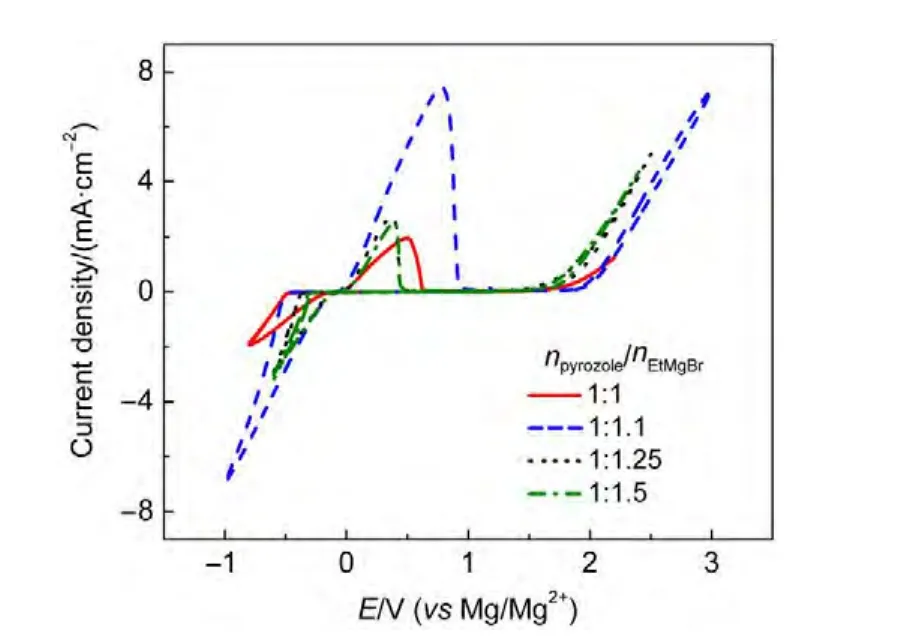
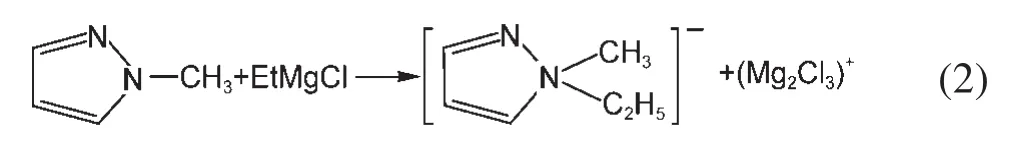
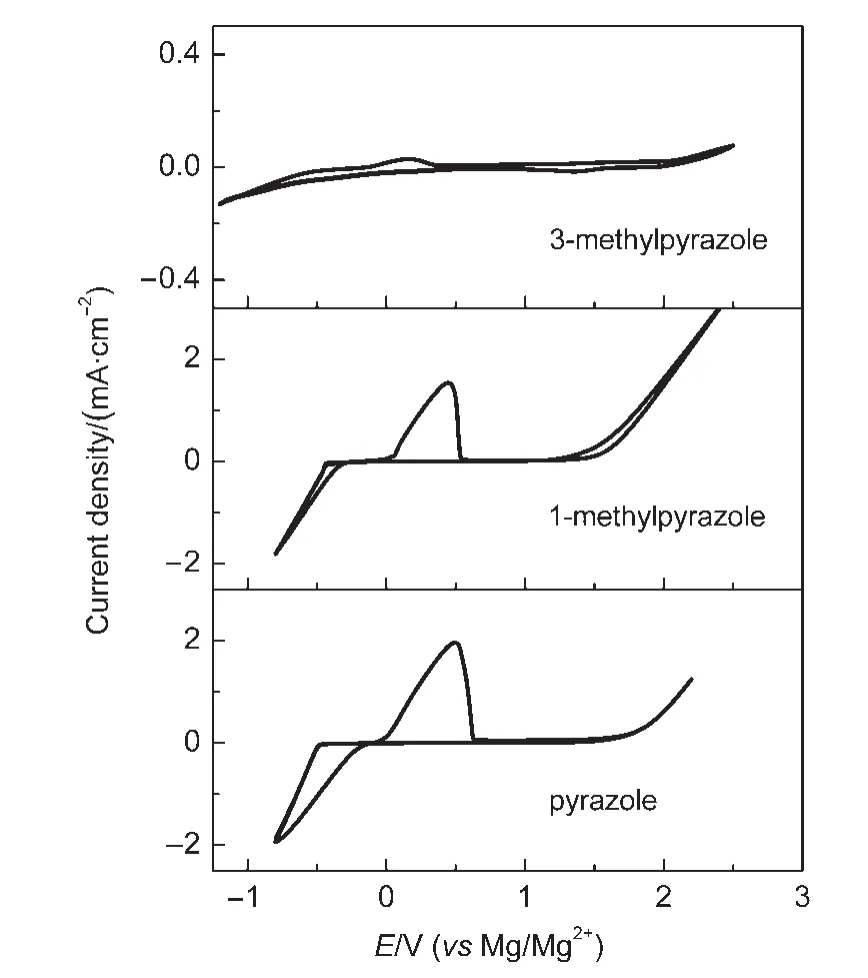
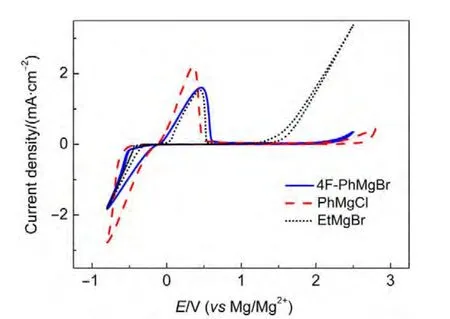
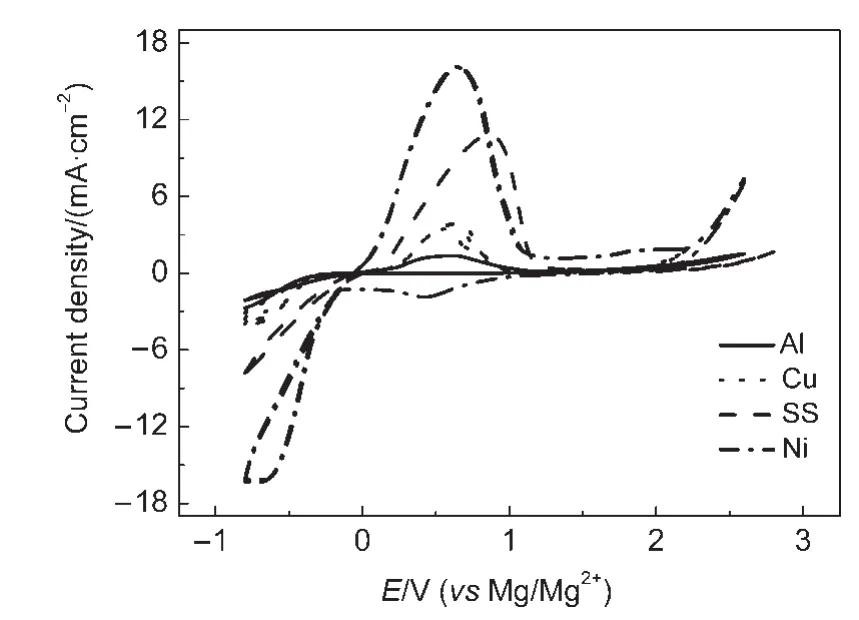
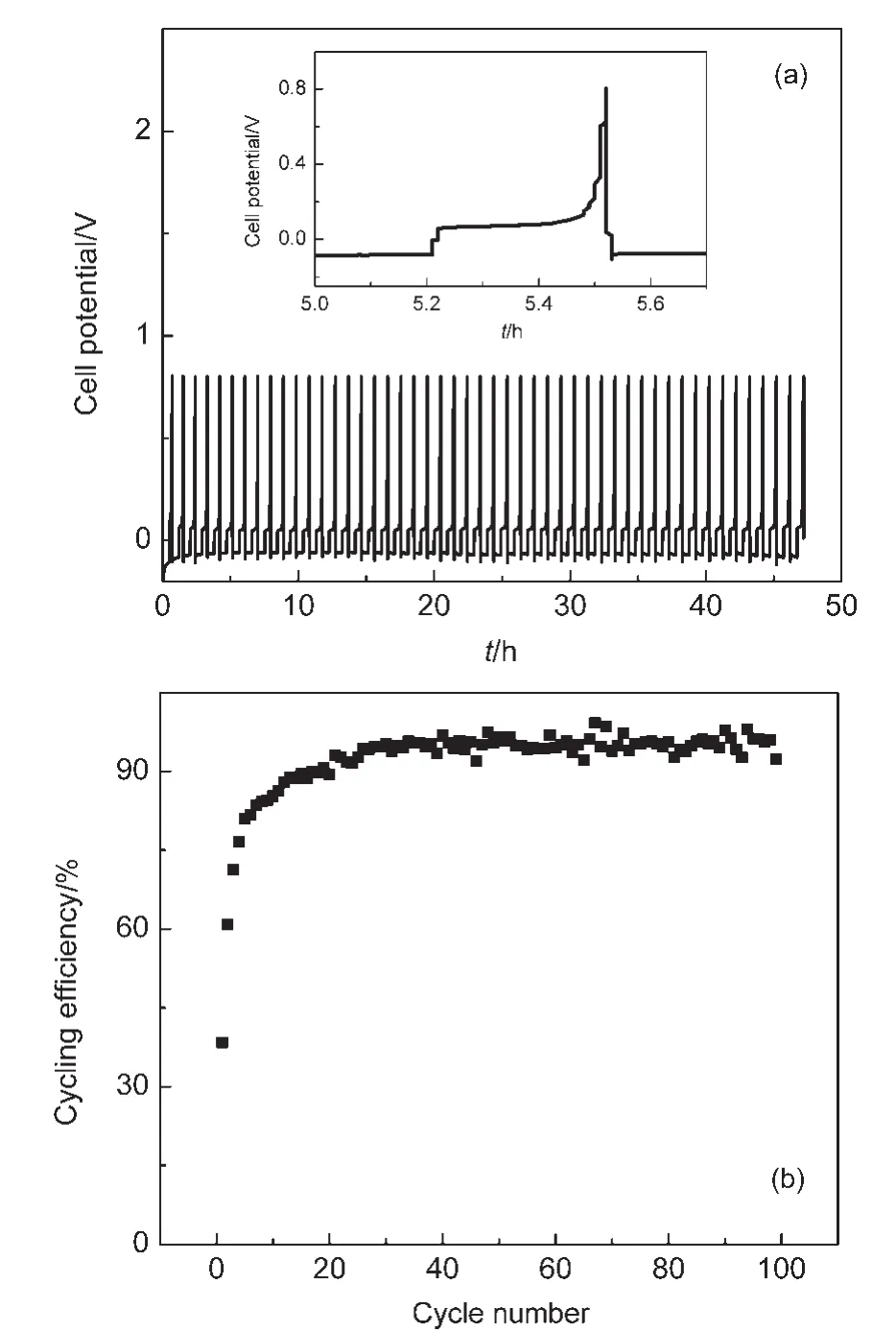
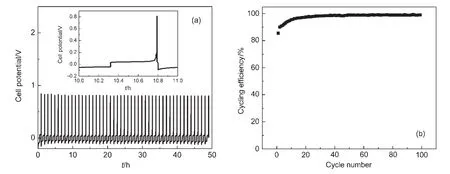
由于鎂的反應活性較高,在大多數電解液中都會形成表面鈍化膜,致使鎂離子無法穿過,從而難以進行可逆的鎂沉積和溶出,限制了其電化學活性.研究表明,在格氏試劑(RMgX,R為烷基或芳基,X為鹵素)、3-6Mg(BR2R′2)2(其中R、R′為烷基或芳基)、7-9Mg(AX4-nRn?R?n?)2絡合物(其中 A=Al、B、Sb、P、As、Fe、Ta等,X=Cl、Br、F,R、R′為烷基或芳基,0 格氏試劑與含有活潑氫的氨基化合物反應可以生成氨基鎂鹵絡合物.1990年,Gregory等8通過N-甲基苯胺、N-乙基苯胺、二苯胺和2,5-二甲基吡咯與乙基氯化鎂的醚溶液反應,制備得到了相應的氨基鎂鹵化物,首次報道了溶于醚中的氨基鎂鹵化物電解液體系用于鎂沉積-溶出的電化學性能.Liebenow等9在2000年又比較了N-甲基苯胺溴化鎂、吡咯基溴化鎂、N,N-二(三甲基硅基)氨基氯化鎂三種氨基鎂鹵化物/醚電解液的性能.其中,N,N-二(三甲基硅基)氨基鎂氯化物中由于存在的Si―N―Si基團離域了N上的電子對,提高了電解液的陽極氧化分解電位.在此基礎上,我們課題組研究了苯酚基鎂鹵化物/醚、19苯硫酚基鎂鹵化物/醚、20含氮雜環的吡咯烷基鎂鹵化物/醚21電解液體系中的鎂沉積-溶出性能,為了提高可充電池電解液的性能,進一步系統化了解含氮雜環基鎂鹵化物/醚電解液的性能,本文研究了不同取代基的吡唑與不同格氏試劑反應配制的吡唑基鎂鹵化物/THF電解液體系,重點介紹了1-甲基吡唑基體系.測試了該電解液體系在不同工作電極上的循環伏安性能和鎂沉積-溶出性能,并分析了沉積物的形貌和組成.1-甲基吡唑基鹵化物電解液體系在非惰性電極不銹鋼上顯示出適宜的氧化分解電位和低的鎂沉積-溶出過電位;簡單的制備過程,高的循環效率及穩定的可逆性使該電解液體系在可充鎂電池中顯示出良好的應用前景. 吡唑購于TCI試劑公司,1-甲基吡唑、4-溴吡唑、4-硝基吡唑、4-甲基吡唑、3-甲基吡唑、乙基溴化鎂的四氫呋喃溶液(EtMgBr/THF,1 mol?L-1),苯基氯化鎂的四氫呋喃溶液(PhMgCl/THF,2 mol?L-1)、苯基溴化鎂的四氫呋喃溶液(PhMgBr/THF,1 mol?L-1)、4-氟苯基溴化鎂的四氫呋喃溶液(4-F-PhMgBr/THF,1 mol?L-1)和金屬鎂條購于上海百靈威化學技術有限公司,四氫呋喃(THF)使用之前經實驗室自組裝設備重蒸純化處理去除微量水.所有試劑放置于氬氣氣氛手套箱(MBRAUN,德國UNILAB)中備用.鉑、銅、鎳、不銹鋼(SS)盤(d=2 mm)電極購于上海仙仁儀器儀表有限公司. 使用CHI660電化學工作站(上海辰華儀器有限公司)作循環伏安掃描測試.LAND-CT2001A系統測試扣式電池中電解液的鎂沉積與溶出循環特性.FE30電導率儀和inLab710電極(Mettler Toledo,Switzerland)測試電解液的電導率.D/max-2200/PC型X射線衍射儀(XRD,日本Rigaku公司)和掃描電子顯微鏡(SEM,FEI SIRION)分析在銅基質表面沉積物的組分和形貌. 2.2.1 電解液配置 在氬氣氣氛手套箱(MBRAUN,德國UNILAB)中,室溫條件下,用電子天平稱取一定量的不同種類吡唑加入到干凈的雙口燒瓶中,用移液槍移取相應量的格氏試劑/THF溶液緩慢加入到雙口燒瓶中,邊加邊攪拌,反應時間為1.5 h.反應過程中,瓶口打開,乙烷氣體及時排出.反應完畢,得到微黃色、深黃色電解液(有些有白色沉淀).反應式(1)為吡唑和EtMgCl的反應,除1-甲基吡唑以外,其它吡唑類與格氏試劑的反應與(1)式類似: 2.2.2 電化學性能的測試 電解液中鎂的電化學沉積-溶出性能通過循環伏安曲線(CV)來測定,測試過程均在室溫、氬氣手套箱中進行.實驗采用三電極體系,工作電極使用前用三氧化鋁粉末進行拋光處理,然后用去離子水及無水乙醇清洗,烘干后使用.參比電極、對電極均采用表面用800Cw砂紙打磨處理干凈的鎂條.測試用的三電極管及塞子均經過烘干處理.測量時,從開路電位開始向負方向掃描,掃描速率為50mV?s-1. 通過CR2016扣式電池的恒電流充放電測試電解液中鎂的電化學沉積-溶出循環性能.扣式電池的組裝在氬氣氣氛手套箱中進行.采用的正極為銅箔(99.99%,直徑為12 mm),負極為鎂條(使用前用800Cw砂紙打磨光亮),隔膜為Entek PE膜;電池組裝后在室溫放4 h后進行測量.充放電過程中的電流密度為0.1 mA?cm-2,鎂的沉積過程通過放電時間控制,而鎂的脫出通過充電極限電位到0.8 V來控制,每次充電與放電過程之間緩沖時間為30 s. 2.2.3 XRD及SEM測試 恒電流條件下進行鎂在金屬Cu基質上的沉積,沉積電量為10.7 C?cm-2.實驗采用兩電極體系,室溫條件下在氬氣氣氛的手套箱中進行測定,儀器設備采用CHI660C電化學工作站;正極為銅片,表面用無水乙醇擦拭干凈;負極為鎂條,使用前用800Cw砂紙打磨光亮. 將沉積好的扣式電池在氬氣氣氛手套箱中打開,Cu片上的沉積樣品用重蒸后的THF溶劑進行沖洗,晾干后密封取出,使用X射線衍射儀(Cu靶)對銅基質上的電沉積物進行XRD檢測,掃描范圍為30°-75°,掃描速率為4.0(°)?min-1. 通過JSM-7401F型場發射掃描電子顯微鏡觀察鎂在銅金屬基質上的沉積形貌.沉積后的樣品在氬氣氣氛手套箱中用THF溶劑進行沖洗,晾干后密封取出進行SEM檢測. 圖1為吡唑(C3H4N2)與EtMgBr/THF按不同摩爾比配制的C3H3N2MgBr/THF電解液在鉑盤電極上的鎂沉積-溶出的循環伏安性能對比,對電極和參比電極為鎂條,掃描速率為50 mV?s-1.在幾種溶液中都可以進行鎂的電化學沉積和溶出.在從開路電位向負向掃描的過程中,從-0.2 V(vsMg/Mg2+)以下開始出現沉積電流,沉積過程中呈現一個典型的由過電位驅動產生晶核及晶核生長的成核環;正向掃描過程中,沉積鎂的溶出從0 V(vsMg/Mg2+)左右開始,在0.35 V(vsMg/Mg2+)以上出現鎂的氧化溶出峰;再向正電壓方向掃描時電流基本保持不變,表明沒有副反應發生,直到電解液發生分解,剛開始出現電流明顯上升的電位即為電解液的氧化分解電位.吡唑與EtMgBr的摩爾配比對其電化學有一定影響.吡唑與EtMgBr的摩爾比為1:1.1時,電解液顯示出最高的氧化分解電位(1.9 V(vsMg/Mg2+))和最高的沉積-溶出峰電流.此濃度下的鎂溶出起始電位最低,但峰電位最高.繼續增加EtMgBr比例,當吡唑與EtMgBr的摩爾比達到1:1.25和1:1.5時,由于過量EtMgBr的存在,大大降低了電解液體系的氧化分解電位(約1.7 V(vsMg/Mg2+)).Aurbach等13,16對不同酸堿比例制得的“一代電解液”和“二代電解液”的循環伏安比較中發現,電解液制備時酸堿組分比例的變化對鎂沉積-溶出的過電位、峰電流和效率以及電解液的陽極穩定性有很大影響.這是由于電解液中存在復雜的化學平衡,酸堿比例的變化改變了溶液中性分子、陰陽離子的組分和強度,導致了其電化學性能的差異.10,13這四種電解液放置過程中均有沉淀出現,固體沉淀的組分為C3H3N2MgBr.其中吡唑與EtMgBr的摩爾比為1:1的溶液與其它三種相比沉淀明顯較少,雖然其鎂沉積-溶出電流最小,但氧化分解電位適當,在接下來的研究中選擇吡唑與EtMgBr的摩爾配比為1:1. 圖1 吡唑與格氏試劑EtMgBr按不同摩爾比反應制得的1 mol?L-1C3H3N2MgBr/THF電解液在鉑盤電極上的鎂沉積-溶出的循環伏安曲線Fig.1 Typical cyclic voltammograms of Mg depositiondissolution on platinum disk electrode in 1 mol?L-1 C3H3N2MgBr/THF electrolyte obtained from the reaction of pyrozole and EtMgBr at different molar ratios 考慮到在吡唑環的不同位置引入不同的吸電子基團和供電子基團以及基團不同的位置可能會對其與格氏試劑的反應過程產生一定影響,可以得到澄清的溶液.實驗中分別將4-溴吡唑、4-硝基吡唑、4-甲基吡唑、3-甲基吡唑、1-甲基吡唑與EtMgBr按1:1摩爾比反應.其中,4-溴吡唑、4-硝基吡唑、4-甲基吡唑與EtMgBr反應之后溶液中存在大量沉淀,3-甲基吡唑、吡唑與EtMgBr反應后存在少量沉淀,而1-甲基吡唑與EtMgBr反應配制的電解液為澄清溶液,由于1-甲基吡唑中不存在可以與格氏試劑發生反應的活潑氫,反應過程和上面提到的反應式(1)不同,可能的反應過程如(2)式: 圖2分別為吡唑、3-甲基吡唑、1-甲基吡唑與EtMgBr按1:1摩爾比反應配制的1 mol?L-1電解液在鉑盤電極上鎂沉積-溶出的循環伏安性能對比.其中3-甲基吡唑與格氏試劑反應配制的電解液體系陽極氧化分解電位較高,但由于溶液中有明顯沉淀存在,導致鎂沉積-溶出電流較小;與略有沉淀的吡唑體系相比,1-甲基吡唑體系雖然陽極氧化分解電位較低,但溶液澄清,鎂沉積-溶出過電位也較低. 圖3為1-甲基吡唑與三種不同的格氏試劑按1:1摩爾比反應配制的1 mol?L-1電解液在鉑盤電極上的鎂沉積-溶出循環伏安性能對比.可以看出,用吸電子能力強的苯基替換乙基、在苯環上引入強吸電子的F元素,可以改善鎂沉積-溶出性能特別是提高電解液的陽極穩定性.其中1-甲基吡唑與PhMgCl反應配制的電解液體系中鎂沉積-溶出的電流密度高于其余兩種電解液,且氧化分解電位可以達到2.5 V(vsMg/Mg2+),相當于目前比較成熟的“一代電解液”0.25 mol?L-1Mg(AlCl2BuEt)2/THF(2.5 V(vsMg/Mg2+)).與“一代電解液”相比,吡唑電解液體系在制備過程中不需要去除原料中溶劑的過程,具有簡單易行的特點. 圖2 吡唑、1-甲基吡唑、3-甲基吡唑與EtMgBr/THF按1:1摩爾比反應配制的1 mol?L-1電解液在鉑盤電極上鎂沉積-溶出的循環伏安曲線Fig.2 Typical cyclic voltammograms of Mg depositiondissolution on platinum disk electrode in 1 mol?L-1 electrolytes obtained from the reaction of pyrazole,1-methylpyrazole,3-methylpyrazole with EtMgBr/THF at 1:1 molar ratio 圖3 1-甲基吡唑體系與三種不同的格氏試劑以1:1摩爾比反應配制的1 mol?L-1電解液在鉑盤電極上的鎂沉積-溶出循環伏安曲線Fig.3 Typical cyclic voltammograms of Mg depositiondissolution on Pt disk electrode in 1 mol?L-1electrolytes obtained from the reaction of 1-methylpyrazole with different Grignard reagents at 1:1 molar ratio 由于金屬鉑的價格較貴,不適合用作電池體系的集流體,我們進一步通過三電極體系的循環伏安測試比較了常用的不同金屬電極Ni、SS、Cu、Al上的1-甲基吡唑-PhMgCl/THF電解液的電化學性能.圖4給出了這四種金屬電極在1 mol?L-1的1-甲基吡唑-PhMgCl(摩爾比1:1)/THF電解液的循環伏安曲線.可以看出在這四種金屬基質上都可以發生鎂的電化學沉積和溶出,比較鎂沉積-溶出電流可看出其性能由好到壞依次為Ni、SS、Cu、Al,但其陽極氧化穩定性大小依次為SS、Ni、Al、Cu,在SS和Ni電極上該電解液的氧化分解電位約為2.4和2.2 V(vsMg/Mg2+).值得注意的是在SS和Ni上的鎂沉積-溶出電流明顯高于Pt電極上的電流值.Feng等22系統研究了在“一代電解液”中鎂的電化學可逆沉積-溶出的金屬基質效應,發現鎂在Ag、Cu、Ni等幾種不同的金屬基質上都可以進行沉積與溶出,但性能卻存在較大差別.如,鎂在Ag上的沉積過程中因為可以形成銀-鎂合金,對鎂的進一步沉積起到了很大的促進作用,從而使得沉積-溶出峰電流和過電位都明顯好于其他幾種金屬基質.1 mol?L-1的1-甲基吡唑-PhMgCl(摩爾比1:1)/THF電解液在不同金屬電極上的循環伏安曲線的差異也源于Mg沉積過程不同的金屬基質效應. 圖4 1 mol?L-11-甲基吡唑-PhMgCl(摩爾比 1:1)/THF電解液在Al、Cu、SS和Ni工作電極上鎂沉積-溶出的循環伏安曲線Fig.4 Typical cyclic voltammograms of Mg depositiondissolution in 1 mol?L-11-methylpyrazole-PhMgCl(at 1:1 molar ratio)/THF electrolyte onAl,Cu,SS,and Ni electrodes 鎂在電解液體系中可逆沉積-溶出的循環過程是表征可充鎂電池電解液電化學性能的一個重要參數,可以通過扣式電池的恒電流循環充放電方法測試.圖5顯示了1 mol?L-11-甲基吡唑-PhMgCl(1:1摩爾比)/THF電解液在Cu基底上鎂的沉積-溶出循環伏安曲線和效率(鎂溶出的電量和鎂沉積的電量之比).由圖中可以看出穩定循環過程中鎂的沉積、溶出電位約為±0.06 V,鎂在Cu基底上的首次沉積-溶出效率較低,但是穩定之后可達95%以上,表現出較高的鎂可逆沉積-溶出循環效率. 圖5 1 mol?L-11-甲基吡唑-PhMgCl(摩爾比 1:1)/THF電解液中Cu基底上的鎂沉積-溶出循環曲線(a)和循環效率(b)Fig.5 Mg deposition-dissolution cycling curves(a)and efficiencies(b)on Cu substrate in 1 mol?L-1 1-methylpyrazole-PhMgCl(at 1:1 molar ratio)/THF electrolyte 圖6分別為1 mol?L-11-甲基吡唑-PhMgCl(摩爾比1:1)/THF電解液中,鎂在Ni基質上的沉積-溶出性能及沉積-溶出效率.圖中顯示,鎂在Ni基質上的沉積-溶出循環穩定,過電位低,首次效率為85.5%,穩定之后循環效率達98%,但在沉積-溶出循環到80次以上過電位逐漸變大,高于Cu基質上的;在Cu和Ni上均能夠穩定循環200次以上.鎂在不同金屬基質上的電化學沉積-溶出循環性能的差異主要源于不同基質上鎂沉積形貌的區別.12相對于疏松、粗糙的鎂沉積物,致密、均勻的沉積層更有利于鎂的溶出,提高鎂沉積-溶出的長期循環性能.研究表明,與Ni相比,鎂在Cu基質上更容易形成均勻、致密的沉積層.9,21 圖7為1 mol?L-11-甲基吡唑-PhMgCl/THF(摩爾比1:1)電解液體系中電沉積物的XRD圖譜,沉積電量10.7 C?cm-2.圖中的衍射峰32.4°、34.3°、36.9°、47.9°、57.3°、62.9°、68.7°和70.2°為金屬鎂的特征峰(JCPDS 35-0821),43.4°、50.5°和 74.1°為銅基底的衍射峰,除此之外沒有別的峰,這說明了沉積物確實是金屬鎂. 圖8(a,b)顯示了不同放大倍數下鎂在銅金屬基質上沉積形貌,沉積電量10.7 C?cm-2.鎂在銅基質上沉積層致密均勻,這對電解液在可充鎂電池中的實際應用十分重要. 圖6 1 mol?L-11-甲基吡唑-PhMgCl(摩爾比1:1)/THF電解液中Ni基底上的鎂沉積-溶出循環曲線(a)和循環效率(b)Fig.6 Mg deposition-dissolution cycling curves(a)and efficiencies(b)on Ni substrate in 1 mol?L-11-methylpyrazole-PhMgCl(at 1:1 molar ratio)/THF electrolyte 圖7 1 mol?L-11-甲基吡唑-PhMgCl(摩爾比 1:1)/THF電解液中在銅基底上沉積物的XRD譜圖Fig.7 XRD pattern of the electrodeposition on Cu substrate in 1 mol?L-11-methylpyrazole-PhMgCl(at 1:1 molar ratio)/THF electrolyte 圖8 1 mol?L-11-甲基吡唑-PhMgCl/THF(摩爾比 1:1)電解液中鎂在銅基質上的沉積形貌SEM圖Fig.8 SEM images of magnesium deposition morphology on copper in 1 mol?L-11-methylpyrazole-PhMgCl(at 1:1 molar ratio)/THF electrolyte 通過簡單的方法制備出一類吡唑基鎂鹵化物的四氫呋喃溶液作為新型可充鎂電池電解液,運用循環伏安、充放電測試對該類電解液中鎂可逆沉積-溶出性能和陽極氧化分解電位進行了系統的測試,研究表明該電解液體系在非惰性電極上具有較高的陽極氧化分解電位,可逆、穩定的鎂沉積-溶出過程,有希望應用于可充鎂電池中. (1) Zhao,Q.S.;NuLi,Y.N.;Guo,Y.S.;Yang,J.;Wang,J.L.Process.Chem.2011,23(8),1599.[趙青松,努麗燕娜,郭永勝,楊 軍,王久林.化學進展,2011,23(8),1599.] (2) Zheng,Y.P.;Nuli,Y.N.;Yang,J.;Chen,Q.;Wang,J.L.Chem.Ind.Eng.Prog.2011,30(5),1025.[鄭育培,努麗燕娜,楊 軍,陳 強,王久林.化工進展,2011,30(5),1025.] (3) Genders,J.D.;Pletcher,D.J.Electroanal.Chem.Interfa.Electrochem.1986,199,93.doi:10.1016/0022-0728(86)87044-9 (4) Liebenow,C.J.J.Appl.Electrochem.1997,27,221.doi:10.1023/A:1018464210084 (5) Lu,Z.;Schechter,A.;Moshkovich,M.;Aurbach,D.J.Electroanal.Chem.1999,466,203.doi:10.1016/S0022-0728(99)00146-1 (6)Aurbach,D.;Moshkovich,M.;Schechter,A.;Turgeman,R.Electrochem.Solid-State Lett.2000,3,31. (7) Muldoon,J.;Bucur,C.B.;Oliver,A.G.;Sugimoto,T.;Matsui,M.;Kim,H.S.;Allred,G.D.;Zajicek,J.;Kotani,Y.Energy Environ.Sci.2012,5,5941.doi:10.1039/c2ee03029b (8) Gregory,T.D.;Hoffman,R.J.;Winterton,R.C.J.Electrochem.Soc.1990,137,775.doi:10.1149/1.2086553 (9) Liebenow,C.;Yang,Z.;Lobitz,P.Electrochem.Commun.2000,2,641.doi:10.1016/S1388-2481(00)00094-1 (10) Mizrahi,O.;Amir,N.;Pollak,E.;Chusid,O.;Marks,V.;Gottlieb,H.;Larush,L.;Zinigrad,E.;Aurbach,D.J.Electrochem.Soc.2008,155,A103. (11)Aurbach,D.;Lu,Z.;Schechter,A.;Gofer,Y.;Gizbar,H.;Turgeman,R.;Cohen,Y.;Moshkovich,M.;Levi,E.Nature2000,407,724.doi:10.1038/35037553 (12)Aurbach,D.;Schechter,A.;Moshkovich,M.;Cohen,Y.J.Electrochem.Soc.2001,148,A1004. (13)Aurbach,D.;Gizbar,H.;Schechter,A.;Chusid,O.;Gottlieb,H.E.;Gofer,Y.;Goldberg,I.J.Electrochem.Soc.2002,149,A115. (14) Gizbar,H.;Vestfrid,Y.;Chusid,O.;Gofer,Y.;Gottlieb,H.E.;Marks,V.;Aurbach,D.Organometallics2004,23,3826.doi:10.1021/om049949a (15) Vestfried,Y.;Chusid,O.;Gofer,Y.;Aped,P.;Aurbach,D.Organometallics2007,26,3130.doi:10.1021/om061076s (16)Pour,N.;Gofer,Y.;Major,D.T.;Aurbach,D.J.Am.Chem.Soc.2011,133,6270.doi:10.1021/ja1098512 (17) Muldoon,J.;Bucur,C.B.;Oliver,A.G.;Sugimoto,T.;Matsui,M.;Kim,H.S.;Allred,G.D.;Zajicekb,J.;Kotanie,Y.Energy Environ Sci.2012,5,5941.doi:10.1039/c2ee03029b (18) Guo,Y.S.;Zhang,F.;Yang,J.;Wang,F.F.;NuLi,Y.N.;Hirano,S.I.Energy Environ.Sci.2012,5,9100.doi:10.1039/c2ee22509c (19) Wang,F.F.;Guo,Y.S.;Yang,J.;NuLi,Y.N.;Hirano,S.I.Chem.Commun.2012,48,10763.doi:10.1039/c2cc35857c (20) Bian,P.W.;Nuli,Y.N.;Zainapuguli;Yang,J.;Wang,J.L.Acta Phys.-Chim.Sin.2014,30(2),311.[卞沛文,努麗燕娜,再娜甫古麗,楊 軍,王久林.物理化學學報,2014,30(2),311.]doi:10.3866/PKU.WHXB201312201 (21)Zhao,Q.S.;NuLi,Y.N.;Guo,Y.S.;Yang,J.;Wang,J.L.Electrochim.Acta2011,56,6530.doi:10.1016/j.electacta.2011.04.114 (22) Feng,Z.Z.;NuLi,Y.N.;Wang,J.L.;Yang,J.J.Electrochem.Soc.2006,153,C689.2 實驗部分
2.1 試劑與儀器
2.2 實驗方法
3 結果與討論


4 結論
