甲基苯丙胺光譜性質(zhì)的密度泛函分析與指認(rèn)
2014-03-02 03:11:05
當(dāng)代化工 2014年1期
關(guān)鍵詞:振動(dòng)
(浙江省臺(tái)州學(xué)院醫(yī)藥化工學(xué)院, 浙江 臺(tái)州316000)
甲基苯丙胺光譜性質(zhì)的密度泛函分析與指認(rèn)
陳凱浩,鐘愛(ài)國(guó)
(浙江省臺(tái)州學(xué)院醫(yī)藥化工學(xué)院, 浙江 臺(tái)州316000)
采用密度泛函理論的DFT/B3LYP/6-311+G(d,p)方法和基組, 對(duì)甲基苯丙胺的UV-Vis光譜,IR光譜, 1HNMR光譜和熒光光譜進(jìn)行了理論模擬和指認(rèn)。自然電荷計(jì)算表明,胺基N和H原子很可能是其發(fā)揮藥理活性的親電和親核反應(yīng)中心。
冰毒;密度泛函理論;電子光譜
甲基苯丙胺(冰毒, C10H15N)是一種人工合成的中樞神經(jīng)興奮劑,是我國(guó)規(guī)定管制的精神藥品。它已逐步取代本世紀(jì)流行的鴉片、海洛因、大麻、冰毒、可卡因等常用毒品,成為21世紀(jì)全球范圍濫用最為廣泛的毒品。1919年日本化學(xué)家A. Ogata將麻黃堿與紅磷及碘還原,首次合成了甲基苯丙胺,并用于治療哮喘和鼻炎。近年境外傳入的“搖頭丸”,也是冰毒的衍生物。趙金濤等[1]對(duì)冰毒的拉曼散射振動(dòng)模式進(jìn)行了研究;姚紅艷等[2]對(duì)鹽酸甲基苯丙胺的核磁共振(NMR)等光譜進(jìn)行了實(shí)驗(yàn)測(cè)定;張金莊[3]利用紅外顯微鏡,直接采集塑料自封袋表面微量毒品的紅外光譜圖,并與毒品標(biāo)準(zhǔn)紅外光譜的吸收峰尤其是特征吸收峰進(jìn)行定性比對(duì)分析。鄭天等[4]建立了應(yīng)用熒光分光光度法測(cè)定繳獲冰毒中甲基苯丙胺的含量的方法。張潤(rùn)生等[5]采用氣相色譜-傅立葉變換紅外光譜聯(lián)用技術(shù),建立了9種苯丙胺類毒品及其衍生物的分析鑒別方法。由于毒物分析結(jié)果常作為法庭評(píng)判依據(jù),所以對(duì)分析結(jié)果的評(píng)定需全面的圖譜數(shù)據(jù)作為支撐材料。本文采用密度泛函理論方法, 對(duì)甲基苯丙胺的分子光譜如紫外-可見(jiàn)吸收,紅外吸收,拉曼吸收,核磁共振吸收以及熒光發(fā)射光譜等進(jìn)行了理論模擬和指認(rèn),取得了與實(shí)驗(yàn)相吻合的結(jié)果。
1 計(jì)算方法
本文對(duì)冰毒分子采用DFT理論的B3LYP方法在6-311+G(d,p)基組水平上進(jìn)行了優(yōu)化計(jì)算。在優(yōu)化得到的穩(wěn)定構(gòu)型基礎(chǔ)上,采用Freq方法進(jìn)行了頻率分析,結(jié)果表明所有簡(jiǎn)諧振動(dòng)頻率全部為正值,表明其計(jì)算結(jié)果是可信的。本項(xiàng)目的全部計(jì)算工作通過(guò)Gaussian 03程序包在PC機(jī)上完成(圖1)。
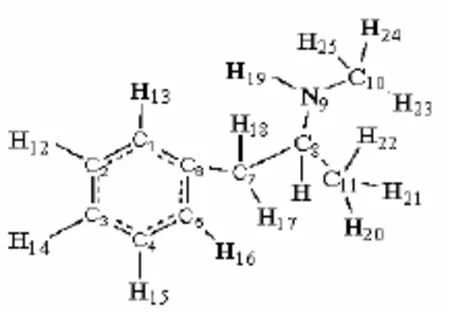
圖 1 甲基苯丙胺(C10H15N)分子結(jié)構(gòu)和原子編號(hào)Fig.1 C10H15N molecular structure and atomic number
2 結(jié)果與討論
2.1 紫外吸收光譜
采用TD DFT/B3LYP/6-311+G(d,p)//DFT/B3LY P/6-311+G(d,p)方法,模擬顯示(圖2),甲基苯丙胺分
子在258 nm (強(qiáng)吸收)和196 nm(弱吸收)處顯示了紫外吸收峰,前者可歸屬于電子從最高占據(jù)軌道(H OMO)躍遷到最低空軌道(LUMO),后者則可歸屬于電子從次高占據(jù)軌道(HOMO-1)躍遷到最低空軌道(LUMO)。這與其標(biāo)準(zhǔn)圖譜顯示甲基苯丙胺分子在2 60 nm處有較強(qiáng)的紫外吸收峰相吻合[6]。

圖2 模擬甲基苯丙胺分子的紫外吸收光譜Fig. 2 Simulation UV absorption spectrum of methamphetamine
2.2 拉曼吸收光譜
拉曼效應(yīng)起源于分子振動(dòng)與轉(zhuǎn)動(dòng), 因此從拉曼光譜中可以得到與紅外分子振動(dòng)互補(bǔ)的信息。采用密度泛函理論DFT/B3LYP方法,在6-311+G(d,p)基組水平上進(jìn)行了分子構(gòu)型優(yōu)化和頻率分析,得到6個(gè)特征峰。對(duì)6個(gè)特征峰的分子振動(dòng)模式進(jìn)行了詳細(xì)的歸屬指認(rèn)。甲基苯丙胺由異丙基和甲胺構(gòu)成的支鏈,取代了苯環(huán)上的一個(gè)氫原子,形成單取代苯類化合物。圖 4是模擬的甲基苯丙胺分子的拉曼散射譜特征峰,主要位于3 450 cm-1(CH3和CH2對(duì)稱、反對(duì)稱譜線), 3 190 cm-1(單取代苯有5個(gè)C-H 伸縮振動(dòng))和1 380 cm-1(CH3和CH2基變形形式的倍頻或合頻), 1 590 cm-1(面內(nèi) C-H 變形衍生的苯環(huán)的振動(dòng))以及690 cm-1(內(nèi)環(huán)變形), 440 cm-1(三角形環(huán)呼吸振動(dòng))范圍內(nèi)。對(duì)比實(shí)驗(yàn)拉曼光譜和理論拉曼光譜,它們的吸收峰基本吻合[7]。
2.3 紅外吸收光譜
物質(zhì)的紅外光譜是其分子結(jié)構(gòu)的反映,譜圖中的吸收峰與分子中各基團(tuán)的振動(dòng)形式相對(duì)應(yīng)(圖3)。采用密度泛函理論方法DFT/B3LYP/6-311+G(d, p)模擬顯示, 冰毒分子在440 cm-1(弱,順式構(gòu)型),690 cm-1(強(qiáng),反式構(gòu)型),1 380 cm-1(弱,甲基的C -H對(duì)稱彎曲振動(dòng)),1 590 cm-1(強(qiáng),苯環(huán)的C-H面外變形振動(dòng)吸收峰) ,3 190 cm-1(中強(qiáng),N-H伸縮振動(dòng)),3 450 cm-1(弱,C-H基的伸縮振動(dòng))等處分別顯示了紅外吸收峰,這些峰與冰毒分子實(shí)驗(yàn)IR圖譜在698.4 cm-1, 1 328.7cm-1, 1 570.7cm-1, 2 93 3.4 cm-1,3 500.8 cm-1有吸收基本吻合[5]。

圖 3 模擬甲基苯丙胺分子的IR圖譜Fig.3 Simulation IR spectrum of methamphetamine

圖4 模擬甲基苯丙胺分子的拉曼圖譜Fig.4 Simulation Raman spectra of methamphetamine
2.4 核磁共振光譜
理論模擬了甲基苯丙胺分子的氫核磁共振譜(見(jiàn)圖5所示),其苯環(huán)上五個(gè)氫(弱,7.12×10-6),主鏈CH2(中,2.52,2.76×10-6); 支鏈CH3(中,1.10× 10-6); 胺基上的CH3(強(qiáng),2.74×10-6);胺基氫(中,2.0 ×10-6) 。這些峰較好地與其實(shí)驗(yàn)值吻合起來(lái)[2],也進(jìn)一步驗(yàn)證了計(jì)算的準(zhǔn)確性。
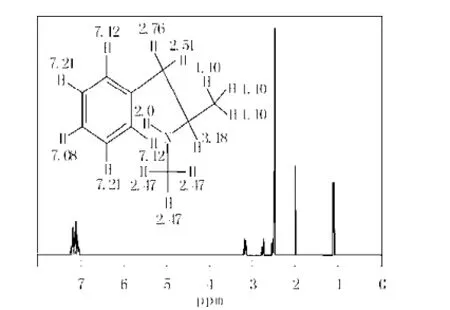
圖5 模擬甲基苯丙胺分子的核磁共振譜Fig.5 Simulation 1HNMR of methamphetamine
2.5 熒光發(fā)射光譜
用TD DFT/B3LYP 6-31+G//CIS HF6-31+g方法,對(duì)標(biāo)題物分子進(jìn)行了分子熒光模擬, 發(fā)現(xiàn)化合物在235.0 nm處的紫外光激勵(lì)下,能產(chǎn)生出弱的熒光發(fā)射強(qiáng)度,理論峰值位于331.5 nm(圖 6,實(shí)驗(yàn)值
為360 nm)[4], 量子化學(xué)模擬計(jì)算發(fā)現(xiàn),該分子的發(fā)光屬于苯環(huán)內(nèi)電子之間的π-π* 躍遷所為。
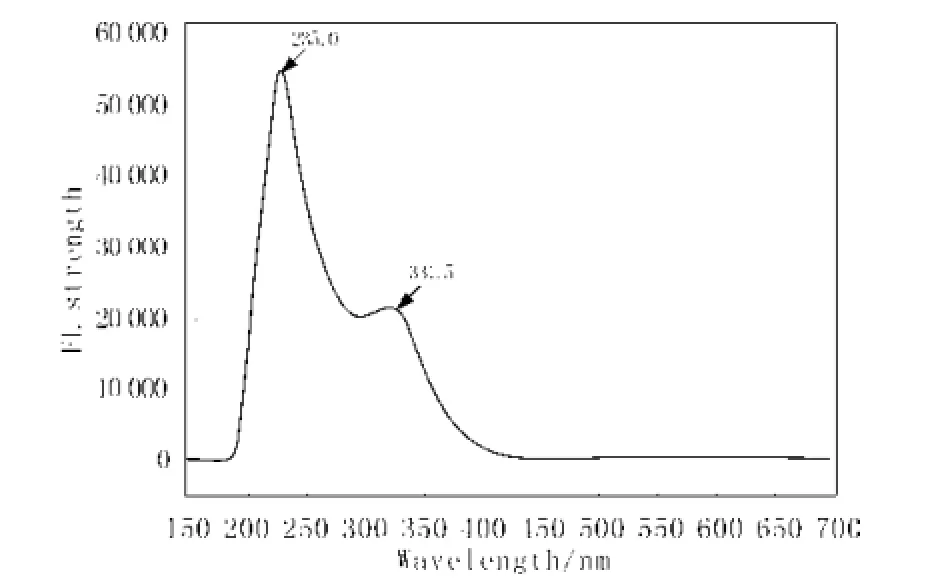
圖6 模擬標(biāo)題物分子熒光Fig. 6 Simulation FL of methamphetamine
2.6 反應(yīng)活性
原子的自然電荷是研究分子的化學(xué)活性點(diǎn)位及確定活性區(qū)域的親核或親電特性強(qiáng)弱的有效方法。表 1的數(shù)據(jù)顯示,胺基氮(N9)電荷最負(fù)(-0.320e),其次為與胺基氮相連的碳(C8,-0.171e);電荷最正的是胺基上的氫原子(H19, 0.12e)。自然電荷計(jì)算表明,胺基(NH)原子很可能是其發(fā)揮藥理和藥理活性的親電和親核反應(yīng)中心。
3 結(jié) 論
對(duì)甲基苯丙胺分子采用密度泛函理論和方法,在6-311+G(d,p)基組水平上進(jìn)行了優(yōu)化計(jì)算。對(duì)其分子光譜(UV-Vis,IR, 1HNMR 以及熒光光譜)進(jìn)行了理論模擬和指認(rèn),取得了與實(shí)驗(yàn)基本吻合的結(jié)果。自然電荷計(jì)算表明,胺基(-NH)氮和氫原子很可能是其發(fā)揮藥理活性的親電和親核反應(yīng)中心。以上理論模擬光譜峰及其指認(rèn)將為甲基苯丙胺分析與檢測(cè)提供幫助。

表1 甲基苯丙胺分子的自然電荷Table 1 Molecular natural charges of methamphetamine
[1]趙金濤, 陳大鵬, 張鵬翔,等. “冰毒”拉曼散射振動(dòng)模式的研究[J].光譜學(xué)與 光譜分析,1999,15(5): 687-690.
[2]姚紅艷,闕玉和,王思宏. 鹽酸甲基安非他命的分析[J].化學(xué)世界,1999, 18:568-570.
[3]張金莊. 基于紅外光譜無(wú)損檢測(cè)自封袋表面微量毒品的研究[J]. 中國(guó)人民公安大學(xué)學(xué)報(bào)(自然科學(xué)版), 2013,75(1):14-16.
[4]鄭天. 熒光分光光度法測(cè)定繳獲冰毒的純度[J]. 江蘇警官學(xué)院學(xué)報(bào), 2013,28(2):103-105.
[5]張潤(rùn)生, 王跨陡, 龔飛君,等. 苯丙胺類毒品及其衍生物的氣相色譜-紅外光譜分析[J]. 分析化學(xué),2012, 40(6):915-919.
[6]傅強(qiáng),廖林川,陳禮莉. HPLC法測(cè)定甲基苯丙胺與苯丙胺[J]. 四川大學(xué)學(xué)報(bào)(醫(yī)學(xué)版), 2007,38(6):l025-1028.
[7]王繼芬, 余靜, 孫興龍, 等. 毒品及其常見(jiàn)添加成分的拉曼光譜快速分析[J]. 光散射學(xué)報(bào), 2012, 24(3):312-315.
Density Functional Analysis and Spectral Identification of Methamphetamine
CHEN Kai-hao,ZHONG Ai-guo
(College of Pharmaceutical and Chemical Engineering, Taizhou University , Zhejiang Taizhou 316000,China)
Using density functional theory DFT/B3LYP/6-311 + G (d, p) method and basis set, UV-Vis spectroscopy, IR spectroscopy,1HNMR spectrum and fluorescence spectroscopy of methamphetamine were studied by the theoretical simulation and identification. Natural charge calculation shows that N and H atoms of amine may be electrophilic and nucleophilic reaction center to play the pharmacological activity.
Ice; Density functional theory; Electronic spectra
O 641
: A
: 1671-0460(2014)01-0029-03
2013-11-18
陳凱浩(1990-),浙江奉化人,臺(tái)州學(xué)院化學(xué)教育專業(yè),研究方向:計(jì)算化學(xué)。E-mail:zhongaiguo@tzc.edu.cn。
鐘愛(ài)國(guó)(1964-),男,研究方向: 計(jì)算化學(xué)。E-mail:xg2268@163.com。
猜你喜歡
科學(xué)大眾(2023年17期)2023-10-26 07:39:14
大電機(jī)技術(shù)(2022年5期)2022-11-17 08:12:48
天天愛(ài)科學(xué)(2020年6期)2020-09-10 07:22:44
瘋狂英語(yǔ)·新讀寫(xiě)(2020年3期)2020-06-06 09:05:56
數(shù)學(xué)物理學(xué)報(bào)(2018年4期)2018-09-14 03:40:58
數(shù)學(xué)物理學(xué)報(bào)(2017年6期)2018-01-22 02:26:40
船海工程(2015年4期)2016-01-05 15:53:26
噪聲與振動(dòng)控制(2015年4期)2015-01-01 07:08:44
計(jì)算物理(2014年2期)2014-03-11 17:01:44
鄭州大學(xué)學(xué)報(bào)(理學(xué)版)(2014年3期)2014-03-01 04:21:00
