多西他賽手性側鏈的制備
2014-03-21 06:26:20王信見夏雪峰姚全興但國蓉
化學與生物工程 2014年4期
王信見,夏雪峰,姚全興,李 靖,但國蓉
(1.重慶泰濠制藥有限公司,重慶400039;2.第三軍醫大學軍事預防醫學院,重慶400038)
多西他賽,英文名docetaxel,化學名稱為2aR-(2aα,4β,4aβ,6β,9α,(αR′,βS′),11α,12α,12aα,12bα)-β-1,1-二甲基乙氧基羰基氨基-α-羰基苯丙酸-12b-乙酰氧-12-苯甲酰氧-2a,3,4,4a,5,6,9,10,11,12,12a,12b-十二氫-4,6,11-三羥基-4a,8,13,13-四甲基-5-氧代-7,11-亞甲基-1 H-環癸五烯并-3,4-苯并-1,2-b-氧雜丁環-9-基酯,是由Sanofi-Aventis公司開發的廣譜抗腫瘤藥,1995年4月在墨西哥上市,目前已在包括中國和歐、美、日等幾十個國家上市。多西他賽2003年的銷售額已高達15.41億美元,上升空間廣闊[1]。
手性側鏈是制備多西他賽的關鍵原料,目前主要的手性側鏈為(3R,4S)-1-tert-butoxycarbonyl-3-(1-ethoxyethoxy)-4-phenyl-2-azetidinone(Ⅰ),其市場供應少,價格昂貴。
文獻報道的制備多西他賽手性側鏈(Ⅰ)的合成方法是:以苯甲醛和對甲氧基苯胺為起始原料,縮合后與乙酰氧基乙酰氯縮合成消旋體四元環,再用硝酸鈰銨氧化脫去對甲氧基苯乙胺后在堿性條件下脫去乙酰基,再用手性試劑拆分得到單一構型,然后與乙烯基乙醚縮合,再與二叔丁基二碳酸酯縮合得到多西他賽手性側鏈。此工藝的特點是拆分步驟發生在第6步,即順式-3-羥基-4-苯基-2-吖叮啶酮的手性拆分。
由于手性拆分需要手性拆分劑、條件苛刻(-70℃),而且拆分工藝靠后導致前面原料的損耗較大[2-3]。因此,作者在文獻[4-5]的基礎上,設計了一條新的合成路線:以苯甲醛(Ⅱ)和S-對甲氧基苯乙胺(Ⅲ)為起始原料,縮合制備N-苯亞甲基-4-甲氧基苯胺(Ⅳ),化合物Ⅳ再與乙酰氧基乙酰氯縮合成四元環,經異丙醚重結晶得到單一手性的(+)-順式-1-對甲氧基苯基-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅴ),再經硝酸鈰銨氧化脫去對甲氧基苯乙胺得到(+)-順式-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅶ),然后在堿性條件下脫去乙酰基得到(+)-順式-3-羥基-4-苯基-2-吖叮啶酮(Ⅷ),最后采用一鍋法,化合物Ⅷ先與乙烯基乙醚縮合、再與二叔丁基二碳酸酯縮合直接得到多西他賽側鏈(Ⅰ)。此路線的特點為:拆分發生在第2步,降低了原料損耗,且該拆分是非手性拆分,不需要手性拆分劑,直接用合適的有機溶劑重結晶即可。具體的合成路線如圖1所示。
1 實驗
1.1 試劑
S-對甲氧基苯乙胺,常州科潤達化工科技有限公司;乙酰氧基乙酰氯,上海嘉辰化工有限公司;硝酸鈰銨、乙烯基乙醚、二叔丁基二碳酸酯、4-二甲胺基吡啶,均為阿拉丁試劑。
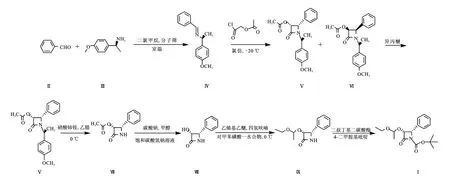
圖1 多西他賽手性側鏈的合成路線Fig.1 The synthetic route of docetaxel chiral side chain
1.2 合成方法
1.2.1 N-苯亞甲基-4-甲氧基苯胺(Ⅳ)的合成
于500mL單口瓶中加入40g苯甲醛(Ⅱ)、170 mL二氯甲烷,攪拌下再加入62g S-對甲氧基苯乙胺(Ⅲ)、40g 4A分子篩,室溫攪拌約24h,液相監控原料基本反應完畢后用硅藻土助濾,用100mL二氯甲烷洗滌,濾液于40℃浴溫旋蒸至無溶劑,得到的油狀物(Ⅳ)直接投入下一步反應,[α]20D=+13.6°(c=0.70,甲醇)。
1.2.2 (+)-順式-1-對甲氧基苯基-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅴ)的合成
于2L三口瓶中加入化合物Ⅳ、500mL氯仿、192 mL三乙胺,攪拌下冷卻至-20℃,滴加乙酰氧基乙酰氯的氯仿溶液(含160mL氯仿,85mL乙酰氧基乙酰氯),約1.5h滴加完畢,轉入室溫攪拌反應直至液相監控化合物Ⅳ反應完畢。傾入500mL水,攪拌10 min后轉入分液漏斗,分出有機相,水相用250mL二氯甲烷提取,合并有機相,用水(500mL×2)洗滌,無水硫酸鈉干燥0.5h,過濾,濾液于40℃旋蒸至干。向殘余物中加入500mL異丙醚加熱溶清,于室溫攪拌過夜析晶,過濾。將濾餅轉入1L單口瓶中,加入500 mL異丙醚,回流溶清后加入2g活性炭,攪拌10min后熱濾,濾液于35℃攪拌析晶2h,抽濾,濾餅于50℃真空干燥,得到干重34.0g的化合物Ⅴ,收率(以化合物Ⅲ計)38.7%,純度95.2%。
1.2.3 (+)-順式-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅶ)的合成
于2L三口瓶中加入34.0g化合物Ⅴ、1 158mL乙腈,室溫攪拌溶清后冷卻至0℃,滴加硝酸鈰銨的水溶液(含170g硝酸鈰銨、430mL水),約1h滴加完畢,繼續于0℃反應約1h直至HPLC監控化合物Ⅴ反應完畢。轉入分液漏斗,傾入1 100mL水,用乙酸乙酯(1 000mL×2)提取,合并有機相,再用5%亞硫酸氫鈉溶液1 000mL和飽和食鹽水1 000mL合并洗滌,無水硫酸鈉干燥2h,過濾。濾液于40℃浴溫濃縮至約150mL,轉入-20℃冰柜放置過夜,過濾,用約200mL正己烷充分洗滌濾餅,于50℃減壓干燥,得到類白色化合物Ⅶ16.8g,收率81.7%,純度97.6%。
1.2.4 (+)-順式-3-羥基-4-苯基-2-吖叮啶酮(Ⅷ)的合成
于1L單口瓶中加入16.0g化合物Ⅶ、610mL甲醇,攪拌下升溫至30℃,加入0.7g碳酸鈉、170mL飽和碳酸氫鈉溶液,繼續于30℃反應約2h直至液相監控化合物Ⅶ反應完畢。抽濾,用100mL乙酸乙酯洗滌,濾液于60℃旋蒸至干,加入750mL乙酸乙酯,于60℃攪拌20min,熱濾,濾液于40℃旋蒸至約70 mL時,室溫攪拌下加入200mL正己烷,30min后抽濾,濾餅于50℃減壓烘干,得到干重約11.5g的化合物Ⅷ,收率90.6%,純度98.7%,[α]20D=+180deg(c=0.65,甲醇)。
1.2.5 多西他賽手性側鏈(Ⅰ)的合成
于1L三口瓶中加入11.5g化合物Ⅷ、550mL四氫呋喃,攪拌下溶畢后冷卻至0℃,加入19mL乙烯基乙醚和2.2g對甲苯磺酸一水合物,25min后加入78mL三乙胺,HPLC檢測化合物Ⅷ反應完畢,加入50g二叔丁基二碳酸酯、2.0g 4-二甲胺基吡啶,室溫反應約2h加入300mL水,用乙酸乙酯(500mL ×3)提取,合并有機相,用飽和食鹽水700mL洗滌,再用無水硫酸鈉干燥2h,過濾,濾液于40℃浴溫濃縮至干。向殘余物中加入700mL正己烷,于回流下攪拌直至基本無固體后加入4g活性炭,攪拌30min,熱濾,用100mL正己烷洗滌,濾液于-20℃冰柜過夜析晶,過濾,濾餅于50℃減壓烘干,得到類白色粉末化合物Ⅰ19.3g,收率81.7%,純度99.7%。
2 結果與討論
2.1 目標化合物表征
多西他賽手性側鏈,[α]20D=+70.4°(c=1.25,CHCl3);1HNMR(250MHz,CDCl3),δ:[0.96(d,J=5.4Hz),1.08(d,J=5.4Hz),3H],[1.09(t,J=7.0 Hz),1.10(t,J=7.0Hz),3H],[1.36(s),1.37(s),9H],[3.23(dq,J=9.5Hz、7.1Hz),3.32(q,J=7.1 Hz),3.65(dq,J=9.5Hz、7.1Hz),2H],[4.48(q,J=5.4Hz),4.69(q,J=5.4Hz),1H],[5.03(d,J=5.8Hz),5.07(d,J=5.8Hz),1H],5.18(d,J=5.8 Hz,1H),7.31(m,5H)。
2.2 討論
1)拆分發生在第2步,對原料的浪費少,且拆分工藝只是簡單的重結晶,無需超低溫條件,也不需要過柱純化,操作簡單。與文獻[2-4]比較,本合成路線的拆分工藝明顯更經濟、更易于工業化。
2)采用一鍋法合成多西他賽手性側鏈,減少了一步后處理,收率達到81.7%。
3)多西他賽手性側鏈合成過程中,后處理極為簡單。正己烷極性小,只有產品能溶解其中,雜質通過簡單的過濾即能一次性除去,產物的純度達到99.5%。
3 結論
以苯甲醛(Ⅱ)和S-對甲氧基苯乙胺(Ⅲ)為起始原料,縮合反應制備N-苯亞甲基-4-甲氧基苯胺(Ⅳ),化合物Ⅳ再與乙酰氧基乙酰氯縮合成四元環,經異丙醚重結晶得到單一手性的(+)-順式-1-對甲氧基苯基-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅴ),再經硝酸鈰銨氧化脫去對甲氧基苯乙胺得到(+)-順式-3-乙酰氧基-4-苯基-2-吖叮啶酮(Ⅶ),然后在堿性條件下脫去乙酰基得到(+)-順式-3-羥基-4-苯基-2-吖叮啶酮(Ⅷ),最后采用一鍋法,化合物Ⅷ先與乙烯基乙醚縮合、再與二叔丁基二碳酸酯縮合直接得到多西他賽手性側鏈(Ⅰ),總收率23.4%。該合成方法反應條件溫和,反應步驟少,收率高。
[1] 馬培奇.多西他賽的臨床地位及其市場趨勢[J].中國醫藥導刊,2006,8(1):53-57.
[2] 烏P,赫爾頓R A.制備多西他賽的方法:中國,101243061A[P].2008-08-13.
[3] 赫頓羅A.紫杉酚中間體的制備方法:中國,1064958C[P].2001-04-25.
[4] JEAN-DOMINIQUE B,ALAIN C.Process for the preparation of β-phenylisoserine andβ-lactam and their analogues:US,5939561A[P].1999-08-17.
[5] OJIMA I,SUN C M,ZUCCO M,et al.A highly efficient route to taxotere by theβ-lactam synthon method[J].Tetrahedron Lett,1993,34(26):4149-4152.
