Performance of Ni/Nano-ZrO2Catalysts for CO Preferential Methanation*
2014-03-25 09:11:13劉其海董新法劉自力
(劉其海)(董新法)(劉自力)**
1School of Chemistry and Chemical Engineering, ZhongKai University of Agriculture and Engineering, Guangzhou 510225, China
2School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China
3School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, China
Performance of Ni/Nano-ZrO2Catalysts for CO Preferential Methanation*
LIU Qihai(劉其海)1, DONG Xinfa(董新法)2and LIU Zili(劉自力)3,**
1School of Chemistry and Chemical Engineering, ZhongKai University of Agriculture and Engineering, Guangzhou 510225, China
2School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China
3School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, China
Large surface areas nano-scale zirconia was prepared by the self-assembly route and was employed as support in nickel catalysts for the CO selective methanation. The effects of Ni loading and the catalyst calcination temperature on the performance of the catalyst for CO selective methanation reaction were investigated. The catalysts were characterized by Brunauer-Emmett-Teller (BET), transmission electron microscope (TEM), X-ray diffraction (XRD) and temperature-programmed reduction (TPR). The results showed that the as-synthesized Ni/nano-ZrO2catalysts presented high activity for CO methanation due to the interaction between Ni active particle and nano zirconia support. The selectivity for the CO methanation influenced significantly by the particle size of the active Ni species. The exorbitant calcination resulted in the conglomeration of dispersive Ni particles and led to the decrease of CO methanation selectivity. Among the catalysts studied, the 7.5% (by mass) Ni/ZrO2catalyst calcinated at 500 °C was the most effective for the CO selective methanation. It can preferentially catalyze the CO methanation with a higher 99% conversion in the CO/CO2competitive methanation system over the temperature range of 260-280 °C, while keeping the CO2conversion relatively low.
selective CO methanation, CO removal, nano zirconia, Ni catalysts
1 INTRODUCTION
Fuel cells have been considered as an effective generator system in recent years for no emission of any pollutant gas [1]. The Proton-exchange Membrane Fuel Cells (PEM FC) are viewed as the most promising candidate for the power sources of small generators and vehicles [2]. PEM FC is fueled by hydrogen-rich gas mixtures usually generated by catalytic reforming of hydrocarbons, alcohols, methane or others followed by CO water gas shift reaction [3-5]. The obtained H2-rich gas inevitably contains 0.5%-2% (by volume) CO, which poisons the PEM FC anode catalyst by strong adsorption of CO and must be removed to a level below 20 ml·m?3[6-8]. Preferential CO oxidation (PROX) by adding air was considered to be a promising method for the deep CO removal from hydrogen-rich gas mixtures [9]. The Au-, Pt-, Ru-, Rhand Cu-based supported catalysts were suggested to be highly active and selective PROX catalyst [10-12]. However, with the presently available catalysts, the undesired oxidation of hydrogen can not be avoided, and an accurate controlling system for the air injecting and the continuous monitoring of the inlet CO concentration were required, which causes additional complications of the PEM FC system. Furthermore, the nitrogen in the added air stream caused unwanted dilution of the hydrogen-rich gas. CO removal from H2-rich gas by selective methanation has been recently proved to be another effective strategy [13]. Compared to PROX, selective CO methanation has the benefit that no air is added to the H2-rich gas stream, and the methane produced is inert to the PEM FC. The amount of hydrogen consumed by the CO methanation is small due to low CO content. The challenge of this method is the presence of relative high CO2volume concentration (20%-25%). If the CO2methanation proceeded, considerable hydrogen loss may occur. Therefore, there is a need to develop a highly active and selective CO methanation catalyst that enables prevention of the CO2methanation.
Recently, high-throughput combinatorial synthesis [14] and screening methods [15] were applied for CO and CO2methanation over zirconia- and ceria-supported noble and base metal catalysts in an attempt to discover new improved catalysts. It was reported that Ni-base and Ru-base catalysts were proved to be effective for selective CO methanation, and the performance of these catalysts could be somewhat improved by using various supports, promoters and preparation procedures [16]. It is considered that the Ni-based catalysts are less promising than the Ru-based catalysts, because they need long pretreatment in reducing atmosphere, possess pyrophoricity and lose rapidly the activity in contact with air. Essential cost-efficiency is the main advantage of the Ni-based systems with respect to Ru-based ones. Consequently, it is important to develop precious-metal-free, highly active and selective catalysts for CO methanation in the presence of CO2. We have suggested the nickel-nano zirconia based catalysts which demonstrate high activity and selectivity for CO methanation in reformate gas [17]. In this work, a number of Ni catalyst with different metal loading supported on nano ZrO2were preparedand studied in the CO selective methanation reaction. Catalytic activity and catalyst characterizations using BET, XRD, TEM, and TPR were discussed.
2 EXPERIMENTAL
2.1 Catalyst preparation
The ZrO2support used in this study was prepared by the self-assembly route using hexadecyltrimethyl ammonium bromide (CTAB). 1.0 g of CTAB dissolved in 20 ml of H2O was added to a 10 mL 0.5 mol·L?1ZrOCl2·8H2O aqueous solution under vigorous stirring and was well mixed to ensure self-assembly. The pH of the mixed solution was adjusted to 9-10 by dropping 25% of ammonia solution to form the zirconium hydroxide sol-gel. The obtained sol-gel precipitate was filtrated after aging for 48 h at 90 °C, washed free of chlorine ions by deionized water and was dried at 110 °C for 12 h. The dried solid was then calcined in the range of 500 °C to 700 °C for 5 h. The obtained sample with surface area of 183 m2·g?1was crushed to powder (≤75 μm) and was employed as the catalyst support.
The ZrO2supported Ni catalysts were prepared by incipient wetness impregnation method. Prior to impregnation, the supports were dried in an oven at 300 °C for 2 h. For the incipient wetness impregnation, 1.1 times the ZrO2volume of the nickel nitrate solution was taken such that it was completely absorbed by the support. By this method, the impregnation was repeated three times for each catalyst and the content of introduced nickel can be accurately controlled. After each impregnation, the wet sample was put in air at room temperature for at least 12 h, then dried at 110 °C for 4 h. After the final impregnation and drying, the sample was calcined at 500-700 °C with a heating rate of 5 °C·min?1for 5 h. The obtained catalyst powders were pressed into pellets (d≈2 mm).
2.2 Catalyst characterization
The real Ni loading of the catalysts was determined by inductively coupled plasma atomic emission spectrometry (ICP, Iris Advantage, ER/S). The specific surface areas (SBET) of the zirconia support and the supported Ni catalysts were determined by BET method using a TriStar II 3020 apparatus. The XRD patterns were recorded using a Purkinje General XD-3 diffractometer using Cu-Kαradiation with a 1.5406 nm of incidence wavelength. The TEM images of the ZrO2support were recorded using a Jeol JEM-100CX II electron microscope (0.14 nm resolution at 200 kV) fitted with an energy-dispersive X-ray spectrometer. The temperature-programmed reduction (H2-TPR) curves were obtained on the AutoChem II 2920 V3.05 instrument (Micromeritics Instrument Co.) at a temperature ramping rate of 10 K·min?1.
2.3 Catalytic reaction
The reaction was performed in a fixed-bed continuous-flow quartz reactor (i.d. ~4 mm, catalyst bed length ~25 mm) at atmospheric pressure, as described in the literature [18-20]. In all experiments, 0.5 g of the catalyst pellets was placed in the reactor. The temperature was measured by a chromel-alumel thermocouple in the middle of the catalyst bed. The experiments were performed with the following gas feed composition (by volume): 1% CO, 70% H2, 3% H2O, 23% CO2, with N2as balance. Total feed flow rate was 3.2 cm3·s?1(STP) to achieve a Mass Hour Space Velocity (MHSV) of 23 000 cm3·g?1·h?1. To prevent the formation of volatile Ni(CO)χ, which is thermodynamically feasible at temperatures <150 °C, the catalytic experiments were performed in the temperature internal of 160-300 °C. Initially, the reactor with catalyst was heated to 160 °C in N2flow, then the reaction mixture was fed to the reactor upon continuous heating to 300 °C; after that the reactor was cooled to 160 °C, catalytic experiments started. The inlet and outlet gas mixtures were analyzed on-line by a gas-chromatograph Agilent 6890 equipped with TDX-01 column and a thermal conductive detector (TCD). The detection limit of CO, CO2, CH4and other gaseous hydrocarbons was ~1 ml·m?3.
3 RESULTS AND DISCUSSION
3.1 Catalyst characterization
The effect of calcination temperature on ZrO2was investigated by TEM as shown in Fig. 1. The TEM images presented the particle size and the surface characteristics of the nano zirconia calcined at 500 °C [Fig. 1 (a)] and 700 °C [Fig. 1 (b)]. The ZrO2support sample calcined at 500 °C comprised of smaller particles (15-20 nm) than that treated at 700 °C (25-35 nm). Additionally, agglomeration of ZrO2particles was observed on the sample calcined at 700 °C, which probably led to the destruction of micropore of the as-synthesized nano ZrO2[21]. Therefore, in this study, the ZrO2calcined at 500 °C was used as the catalyst support. Exorbitant temperature resulting in the decrease of ZrO2surface areas was further proved by the XRD patterns, as shown in Fig. 2. It can be seen that no evident X-ray diffraction peak for NiO species was observed on the XRD patterns of the catalyst calcined at 500 °C. However, when the calcination temperature was increased to 600 °C, a week diffraction peak for NiO at 2θ=43° was found, which became evident as the calcination temperature was increased to 700 °C.
Table 1 presents real Ni loading and specific surface areas of the ZrO2and the Ni/ZrO2catalysts. The real Ni loading was close to the calculated one for all samples. SBETmonotonically decreased from 183 m2·g?1(for ZrO2support) to 64 m2·g?1(for 20Ni/ZrO2) withthe increasing Ni loading. This change was probably due to the micro-Ni particles blocking the pores of ZrO2support [22].

Figure 1 TEM images (×104) of the nano ZrO2calcinated at 500 °C (a) and 700 °C (b)
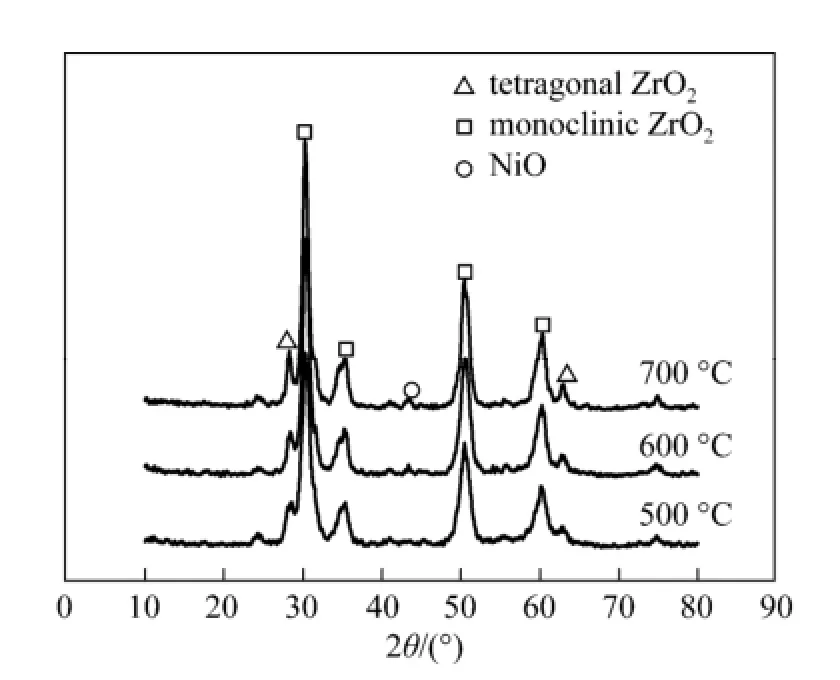
Figure 2 XRD patterns of 4Ni/ZrO2catalysts calcinated at different temperature

Table 1 SBETof the catalysts employed in the present study
Figure 3 showed the XRD patterns of the nano-ZrO2and the Ni/ZrO2catalysts. It was found that the XRD patterns of the catalysts with Ni loading below 7.5% (by mass) were the same as the pattern of ZrO2, which demonstrated neither nickel oxide, nor metallic nickel peaks. This indicated that the nickel species in these catalysts were in finely dispersed state. As the Ni mass loading was increased to higher 13.7% (15Ni/ZrO2, 20Ni/ZrO2catalysts), the peaks corresponding to nickel oxide appeared in the XRD patterns (see Fig. 3), indicating the conglomeration of the Ni particles.

Figure 3 XRD patterns of the ZrO2and the as-synthesized 2Ni/ZrO2, 4Ni/ZrO2, 8Ni/ZrO2, 15Ni/ZrO2and 20Ni/ZrO2catalysts
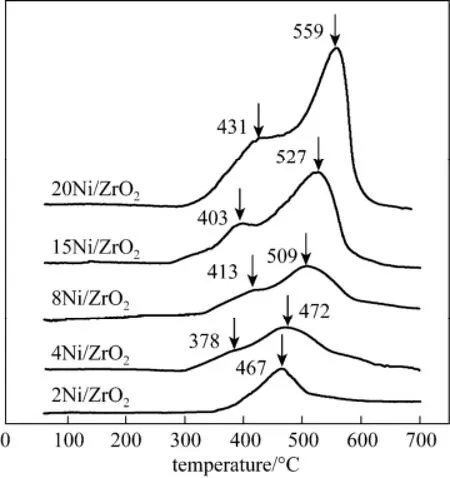
Figure 4 TPR profiles of the 2Ni/ZrO2, 4Ni/ZrO2, 8Ni/ZrO2, 15Ni/ZrO2and 20Ni/ZrO2catalysts
The Ni/ZrO2catalysts reducibility and the NiO dispersibility on the ZrO2support were investigated through TPR experiments. The TPR results of the catalysts were shown in Fig. 4. Since ZrO2did not show any reduction peak under 700 °C, it was assumed that all the consumed hydrogen was contributed to the reduction of NiO. Over the 2Ni/ZrO2catalyst, only a broad peak at around 467 °C was observed. With Ni mass loading increasing to higher 3.7% (4Ni/ZrO2catalyst), a shoulder peak at lower-temperature was found. According to the XRD data, these shoulder peaks were likely due to the reduction of the bulk NiO species. As Ni loading increasing, these shoulder peaks gradually became evident and shifted to higher temperature, indicating the increase of the unsupported bulk NiO species in the catalyst. These bulk NiO species hardly interacted with the ZrO2support, and was detrimental to the CO methanation selectivity [15]. Except for the shoulder peak, the peaks at higher temperature were associated to the reduction of the NiO species that finely dispersing on the ZrO2surface. With Ni loading increasing, both the intensity and the temperature of these peaks increased, indicating the increase of the dispersed Ni species. These dispersed Ni particles, which strongly bonded with ZrO2support, was reported to correlate significantly with the catalytic selectivity for CO methanation [17].
3.2 Catalytic performance
3.2.1 Effect of Ni loading
Figure 5 presents the temperature dependencies of the CO concentration and CO2conversion at selective CO methanation over 2Ni/ZrO2, 4Ni/ZrO2, 8Ni/ZrO215Ni/ZrO2and 20Ni/ZrO2catalysts. Of all catalysts, with the increasing temperature, the CO conversion first increased dramatically, reached a maximum and then slightly decreased. They are typical for the CO selective methanation at various temperature [18-20], because the CO2methanation gradually predominate in the competitive reaction system with the increasing temperature. It was also found that as the Ni loading increased, the temperature dependencies of the CO methnation reaction shifted to the lower temperature region. The most profound shift was observed from the 2Ni/ZrO2catalyst to 4Ni/ZrO2catalyst, which presented around 40 °C excursion to lower temperature at the same CO conversion. The high-loaded nickel catalysts (8Ni/ZrO2, 15Ni/ZrO2and 20/NiZrO2) demonstrated similar temperature dependencies of the CO methanation. In the temperature interval of 180-240 °C, CO methanation preferentially proceeded over the catalysts, and high selectivity of CO methanation was obtained. At higher 260 °C reaction temperature, the CO methanation selectivity slightly decreased and, probably, the Reverse Water Gas Shift (RWGS) reaction proceed in parallel with the CO methanation [23, 24]. For the CO2methanation reaction, they occurred only noticeably at temperatures above 220 °C. As the temperature increased to higher 220 °C, the CO2methanation proceeded rapidly, which resulted in the decrease of the CO methanation selectivity.
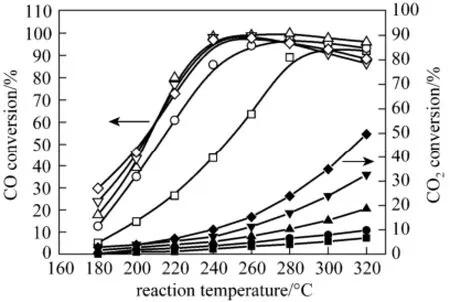
Figure 5 Effect of Ni loading of the Ni/ZrO2catalyst on the CO conversion(open) and the CO2conversion (solid)□, ■ 2Ni/ZrO2; ○, ● 4Ni/ZrO2; △, ▲ 8Ni/ZrO2;▽, ▼ 15Ni/ZrO2; ◇, ◆ 20Ni/ZrO2
The active Ni surface area (SNi) and the dispersibility of NiO over the ZrO2surface represented probable reasons for these observations. Likely, the dispersive nickel surface area increased drastically with Ni loading increasing. In low Ni mass loading catalysts (<7.5%), nickel distributed uniformly over zirconia surface as microcrystal. With loading up, the number of these Ni crystallites obviously increases that lead to the increase of SNiand catalytic activity. At the same time, clusters of Ni species were formed. In the highloaded catalysts (15Ni/ZrO2and 20Ni/ZrO2), contribution of Ni cluster to SNiseems to be insignificant for the CO methanation activities. The higher activity and lower selectivity of the catalysts with high nickel loading as compared to the low-loaded catalysts can be explained by that the CO2methanation reaction occurred predominantly on the clustered nickel sites, in consistence with the results in [16]. In that work, considerable increase in the conversion of CO2into CH4was observed with increasing size of Ni active particles. As Fig. 5 shows, among the catalysts studied, 8Ni/ZrO2is the most effective for CO selective methanation. This catalyst provided the CO conversion higher than 98% in the widest temperature window of 240-280 °C with a relatively low CO2conversion (<11%).
3.2.2 Effect of calcination
Figure 6 presented the effect of calcination temperature of the 8Ni/ZrO2catalyst on CO conversion and CO2conversion at the CO/CO2competitive methanation system. It was seen that the 8Ni/ZrO2catalyst calcined at 500 °C showed the optimum performance for selective CO methanation in reformate gas. With the increase of the calcination from 500 °C to 700 °C, the catalytic activity for the CO methanation decreased, but for the CO2methanation increased significantly. The reason for this observation may be due to the destruction of ZrO2pores and the aggregation of dispersed NiO particles when they was calcined at excessively high temperature. In 8Ni/ZrO2calcined at 500 °C, the nickel species was finely dispersed, which can also be reduced easily to metallic state. Exactly, this was likely responsible for high activity and selectivity of the catalyst for CO selective methanation reaction [14].
4 CONCLUSIONS
The performance of the nanoscale zirconia supported Ni catalysts [1.9%-18.6% (by mass) Ni/ZrO2] for selective CO methanation in a reformate gas was investigated. The synthesized zirconia-supported Ni catalysts presented high activity and selectivity for CO methanation. Among the catalysts studied, the 7.5% (by mass) Ni/ZrO2catalyst calcined at 500 °C was the most effective for the CO selective methanation in the CO/CO2competitive methanation system. This catalyst provided a higher than 99% of CO conversion in the CO/CO2competitive methanation reactions over the temperature interval of 260-280 °C, while keeping CO2conversion relatively low.
REFERENCES
1 Costamagna, P., Srinivasan, S., “Quantum jumps in the PEMFC science and technology from the 1960s to the year 2000: Part II. Engineering, technology development and application aspects”, J. Power Sources, 102 (1-2), 253-269 (2001).
2 Song, S.Q., Tsiakaras, P., “Recent progress in direct ethanol proton exchange membrane fuel cells (DE-PEMFCs)”, Appl. Catal. B, 63 (3-4), 187-193 (2006).
3 Liu, Q.B., Hong, H., Yuan, J.L., “Experimental investigation of hydrogen production integrated methanol steam reforming with middle-temperature solar thermal energy”, Appl. Energy, 86 (2), 155-162 (2009).
4 Rabe, S., Vogel, F.., “A thermogravimetric study of the partial oxidation of methanol for hydrogen production over a Cu/ZnO/Al2O3catalyst”, Appl. Catal. B, 84 (3-4), 827-834 (2008).
5 Huang, T.J., Huang, M.C., “Effect of Ni content on hydrogen production via steam reforming of methane over Ni/GDC catalysts”, Chem. Eng. J., 145 (1), 149-153 (2008).
6 Gamburzev, S., Appleby, A.J., “Recent progress in performance improvement of the proton exchange membrane fuel cell (PEMFC)”, J. Power Sources, 107 (1), 5-12 (2002).
7 David, T., ?nsan, Z., “Onboard fuel conversion for hydrogen-fuel-cell-driven vehicles”, Catal. Rev., 43 (1-2), 31-84 (2001).
8 Huang, C.P., Jiang, R.C., Elbaccouch, M., Muradov, N., Fenton, J.M.,“On-board removal of CO and other impurities in hydrogen for PEM fuel cell applications”, J. Power Sources, 162 (1), 563-571 (2006).
9 Trimm, D.L., “Minimisation of carbon monoxide in a hydrogen stream for fuel cell application”, Appl. Catal. A, 296 (1), 1-11 (2005).
10 Zou, H.B., Dong, X.F., Lin, W.M., “Selective CO oxidation in hydrogen-rich gas over CuO/CeO2catalysts”, Appl. Surf. Sci., 253 (5), 2893-2898 (2006).
11 Chen, X.R., Zou, H.B., Chen, S.Z., “Selective oxidation of CO in excess H2over Ru/Al2O3catalysts modified with metal oxide”, J. Natural Gas Chemistry, 16 (4), 409-414 (2007).
12 Huang, Y.Q., Wang, A.Q., Li, L., “Ir-in-ceria: A highly selective catalyst for preferential CO oxidation”, J Catal., 255 (2), 144-152 (2008).
13 Dagle, R.A., Wang, Y., Xia, G., “Selective CO methanation catalysts for fuel processing applications”, Appl. Catal. A, 326 (2), 213-218 (2007).
14 Kr?mer, M., Duisberg, M., St?we, K., “Highly selective CO methanation catalysts for the purification of hydrogen-rich gas mixtures”, J. Catal., 251 (2), 410-422 (2007).
15 Yaccato, K., Carhart, R., Hagemeyer, A., “Competitive CO and CO2methanation over supported noble metal catalysts in high throughput scanning mass spectrometer”, Appl. Catal. A, 296 (1), 30-48 (2005).
16 Takenaka, S., Shimizu, T., Otsuka, K., “Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts”, Inter. J. Hydrogen Energy, 29 (10), 1065-1073 (2004).
17 Liu, Q.H., Dong, X.F., Mo, X.M., “Selective catalytic methanation of CO in hydrogen-rich gases over Ni/ZrO2catalyst”, J. Natural Gas Chem., 17 (3), 268-272 (2008).
18 Liu, Q.H., Dong, X.F., Song, Y.B., Lin, W.M., “Removal of CO from reformed fuels by selective methanation over amorphous Ni-B-Zr-Oδcatalysts”, J. Natural Gas Chem., 18 (2), 173-178 (2009).
19 Liu, Q.H., Liu, Z.L., Liao, L.W., Dong, X.F., “Selective CO methanation over amorphous Ni-Ru-B/ZrO2catalyst for hydrogen-rich gas purification”, J. Natural Gas Chem., 19 (5), 497-502 (2010).
20 Liu, Q.H., Dong, X.F., Lin, W.M., “Highly selective CO methanation over amorphous Ni-Ru-B/ZrO2catalyst”, Chin. Chem. Lett., 20 (8), 889-892 (2009).
21 Kester, B.K., Zagli, E., Falconer, L.J., “Methanation of carbon monoxide and carbon dioxide on Ni/Al2O3catalysts: Effects of nickel loading”, J. Catal., 22 (2), 311-319 (1986).
22 Batista, M.S., Santiago, E.I., Assaf, E.M., “Evaluation of the water-gas shift and CO methanation processes for purification of reformate gases and the coupling to a PEM fuel cell system”, J. Power Sources, 145 (1), 50-54 (2005).
23 Kim, S.H., Nam, S., Lim, T., “Effect of pretreatment on the activity of Ni catalyst for CO removal reaction by water-gas shift and methanation”, Appl. Catal. B, 81 (1-2), 97-104 (2008).
24 Liu, Q.H., Liao, L.W., Liu, Z.L., Dong, X.F., “Effect of ZrO2crystalline phase on the performance of Ni-B/ZrO2catalyst for the CO selective methanation”, Chin. J. Chem. Eng., 19 (3), 434-438 (2011).
Received 2012-03-12, accepted 2012-07-09.
* Supported by the National Natural Science Foundation of China (21276054, 21376280).
** To whom correspondence should be addressed. E-mail: gzdxlzl@163.com
 Chinese Journal of Chemical Engineering2014年2期
Chinese Journal of Chemical Engineering2014年2期
- Chinese Journal of Chemical Engineering的其它文章
- Kinetics of Glucose Ethanolysis Catalyzed by Extremely Low Sulfuric Acid in Ethanol Medium*
- Synthesis of Sub-micrometer Lithium Iron Phosphate Particles for Lithium Ion Battery by Using Supercritical Hydrothermal Method
- Hydrogenation of Silicon Tetrachloride in Microwave Plasma
- Effects of Solvent and Impurities on Crystal Morphology of Zinc Lactate Trihydrate*
- Large-eddy Simulation of Ethanol Spray-Air Combustion and Its Experimental Validation*
- Kinetic and Thermodynamic Studies of Acid Scarlet 3R Adsorption onto Low-cost Adsorbent Developed from Sludge and Straw*