基于低溫醇水體系的均相沉淀法制備納米FePO4
2014-04-23 02:22:44伍麗萍鄭典模田澤由黃同林李海港
電源技術 2014年1期
伍麗萍, 鄭典模,田澤由,黃同林,李海港
(1.江西省工業安全工程技術研究中心,江西南昌 330030;2.南昌大學環境與化學工程學院,江西南昌 330031;3.江西省化學工業設計院,江西南昌 330030)
隨著環境保護和電動汽車技術等領域的發展,對安全、高效、價格低廉的新能源需求日益增加。LiFePO4作為鋰離子電池的正極材料,具有原料豐富、價格低廉、放電比容量大(170mAh/g)[1]、無毒性、環境友好等優點。目前,鋰離子電池使用的LiFePO4粉體粒徑一般在5μm左右,較大顆粒的LiFePO4粉體中鋰離子擴散困難,擴散系數低,導致鋰離子電池的電化學性能不夠理想。超細LiFePO4可以縮短鋰離子的擴散路徑,提高其擴散系數,納米化的LiFePO4可一定程度上抑制正極材料發生不可逆相變,大為改善鋰離子電池的電化學性能[2-4]。FePO4是制備LiFePO4的骨架材料,其制備的LiFePO4顆粒比FePO4大,只有超細(納米)尺度的FePO4才可能制備超細(納米)的 LiFePO4。
傳統液相法制備納米粉體的普遍性問題是粒徑尺寸難控制、顆粒易團聚,將醇水體系應用到液相法制備納米粉體,可提高粉體分散性和均勻性[5-6]。本文探討在低溫醇水體系中制備納米FePO4粉體的工藝。
1 實驗材料和方法
1.1 試劑和儀器
試劑:七水合硫酸亞鐵(FeSO4·7 H2O,AR),硫酸(H2SO4,AR),磷酸(H3PO4,AR),雙氧水(H2O2,AR),氨水(NH3,AR),SDS(AR),無水乙醇(AR),蒸餾水等。實驗采用傅里葉紅外光譜儀(FTIR,FT-IR360,NICOLET)進行樣品成分分析,采用場發射掃描電子顯微鏡(FESEM,HITACHI,S4800,加速電壓 30 kV)檢測樣品的形貌和粒度,采用粉末X射線衍射儀(XRD、D8 FOCUS、AXS)進行樣品晶型分析,采用粒度分布儀進行樣品粒度分布分析。
1.2 實驗方法和條件
制備FePO4反應如下:
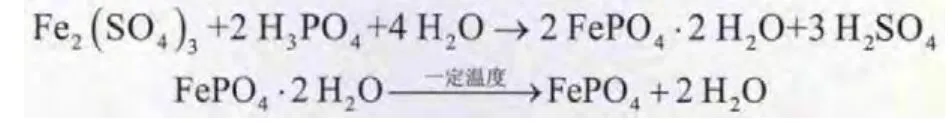
將適量FeSO4·7 H2O溶解于去離子水中,加入適量H2SO4和H2O2,配制成一定濃度的Fe2(SO4)3溶液,同時配制同等摩爾濃度的H3PO4溶液。取一定體積無水乙醇和表面活性劑于反應器內攪拌混合均勻,同時以5m L/min加入Fe2(SO4)3溶液和H3PO4溶液,反應一定時間后,用氨水調節pH至1.9±0.5,陳化1 h后進行抽濾,用去離子水和無水乙醇洗滌抽濾,進行噴霧干燥后于450℃煅燒2 h,即得到FePO4粉體。
實驗方案流程圖如圖1所示。
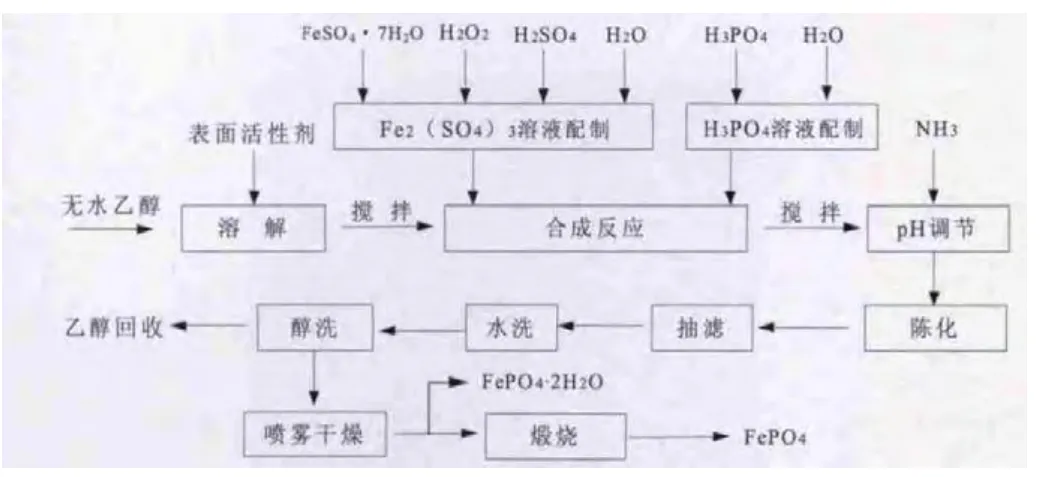
圖1 實驗流程圖
2 實驗結果的討論
實驗考察了醇水比例、反應物濃度、攪拌速度和反應時間對FePO4粉體粒度的影響。
2.1 實驗
2.1.1 醇水比例對FePO4平均粒徑影響
在反應物濃度為0.4mol/L,攪拌速度為3 000 r/m in,反應溫度為35℃,反應時間為1 h的條件下,考察醇水比例為1∶8、1∶4、1∶2、1∶1、2∶1 時醇水比例對 FePO4粉體粒度的影響。實驗結果如圖2所示,當醇水比例<1時,粉體粒度隨著醇水比例增加顯著降低,而當醇水比例≥1后,粉體粒度隨醇水比例增大而基本不變,因此醇水比例取1∶1比較合適。

圖2 醇水比例與平均粒度的關系
文獻[7]報道醇水體系中,其介電常數隨乙醇加入量增加而減小,醇水體系中的反應產物的顆粒粒徑隨之減小。對于水溶液體系而言,若生成的沉淀粒子顆粒尺寸小,比表面積大,就很容易形成團聚體,加入乙醇后,醇水可以無限互溶,使得顆粒表面吸附的水分子大量替換為醇分子,降低了顆粒的表面張力和表面能,從而有效減輕了顆粒間的團聚趨勢。另外,乙醇分子大于水分子,顆粒表面的乙醇分子能起到一定的空間位阻作用[8],降低顆粒間碰撞的幾率,利于生成尺寸小、分散性好的顆粒。
2.1.2 攪拌速度對FePO4平均粒徑影響
在醇水比例為1∶1,反應物濃度為0.4mol/L,反應溫度為35℃,反應時間為1 h的條件下,考察了攪拌速度為100、500、1 000、2 000、3 000 r/min 時攪拌速度對 FePO4粉體粒度的影響。實驗結果如圖3所示,當轉速≤2 000 r/min時,FePO4粉體粒度隨著攪拌速度增大而增大;當轉速>2 000 r/m in,繼續增大轉速,FePO4粉體粒度呈現減小的趨勢。

圖3 攪拌速度與平均粒度的關系
FePO4粉體粒度隨著攪拌速度的增大呈現先增大后減小的趨勢,其原因是由于初始時攪拌速度的增大加劇了顆粒之間的相互碰撞,使團聚加劇,形成了較大的團聚體顆粒;隨著攪拌速度的增大,流體中的渦流剪切力隨之增大,團聚體的破碎機率也增大,因此團聚體不會無限增大,最終在某個攪拌速度出現一個最大值,繼續增大攪拌速度,顆粒將會隨之減小[9]。
2.1.3 反應物濃度對FePO4平均粒度影響
在醇水比例為1∶1,攪拌速度為3 000 r/min,反應溫度為35℃,反應時間為1 h,的條件下,考察了反應物濃度為0.2、0.4、0.6、0.8、1.2mol/L 時反應物濃度對 FePO4粉體制備的影響。
從圖4可見,隨著反應物濃度的增加,FePO4粉體粒徑呈先增大后減小趨勢。根據結晶過程原理,結晶過程包括晶核形成和晶核生長過程,在較低濃度時,隨著反應物濃度的增大,反應中形成的晶核數目增加,晶核之間的接觸概率增大,核化速率加快,導致晶粒增大;但是當達到一定的反應物濃度時,體系能迅速形成大量的晶核,單個晶核所能得到的反應生成的結晶物少,較難形成較大的晶粒[9]。
2.1.4 反應時間對FePO4平均粒徑影響
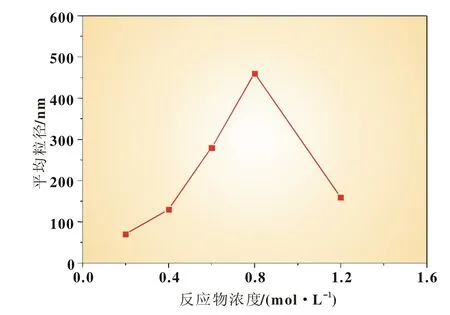
圖4 反應物濃度與平均粒徑的關系
在醇水比例為1∶1,反應物濃度為0.4mol/L,反應溫度為35℃,反應時間為1 h的條件下,考察了反應時間為0.5、1、2、4、8 h時反應時間對FePO4粉體粒度的影響。實驗發現,當反應時間在0~8 h,隨著反應時間增加FePO4粉體粒度呈增大趨勢,如圖5所示。
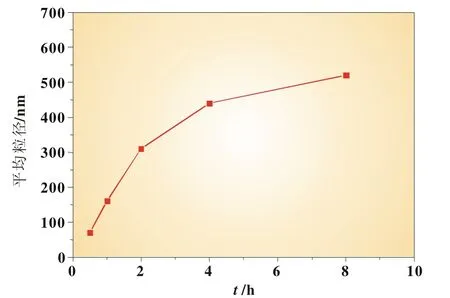
圖5 反應時間與平均粒徑的關系
根據結晶原理,結晶過程包括晶核的形成和生長過程,隨著反應時間的增加,反應初期形成的大量晶核的相互碰撞團聚機會增大,晶核生長的時間更長,易形成更大的顆粒[9]。根據實驗數據,反應時間0.5 h最佳,其反應已經徹底完成,且可得到較細的反應產物。
2.2 樣品表征
在醇水比例為1∶1,反應物濃度為0.2mol/L,攪拌速度為3 000 r/min,反應時間為0.5 h,反應溫度為35℃條件下制得FePO4·2 H2O樣品,將所得FePO4·2H2O樣品于450℃煅燒2 h制得FePO4樣品,對樣品進行FITR、XRD和FESEM檢測分析。
2.2.1 FePO4·2H2O樣品的紅外光譜分析(FTIR)測定
圖6是FePO4·2 H2O樣品的紅外吸收光譜圖。FePO4·2 H2O的光譜活性主要由結晶水和磷酸根基團引起[10]。其中PO43-的吸收帶主要有兩個,即位于1 120~940 cm-1區間的強吸收峰和650~540 cm-1區間的中強吸收峰;結晶水的主要吸收能帶3 650~3 100 cm-1區間的伸縮振動和1 800~1 500 cm-1區間的彎曲振動。圖6的樣品紅外光譜中3 388 cm-1和1 636 cm-1分別是所帶結合水(或者游離水)的伸縮振動(νOH)及彎曲振動(δOH);1 117 cm-1和 982 cm-1分別屬于 PO43-的 ν3(F2)(即PO反對稱伸縮振動)和ν1(A1)(即PO對稱伸縮振動);539 cm-1的中強吸收峰為 PO43-的 ν4(F2)(即 PO2反對稱彎曲振動)。
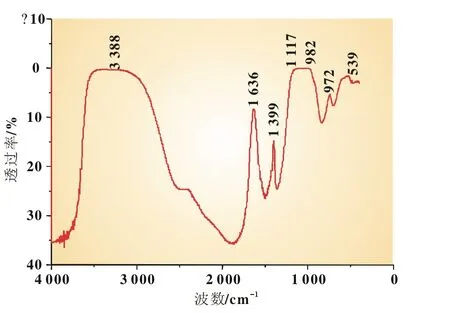
圖6 FePO4·2 H2O的紅外譜圖
2.2.2 FePO4·2H2O樣品的場發射掃描電子顯微鏡測定
由圖7可見,所制得的FePO4·2 H2O樣品呈不定形態,顆粒分散比較均勻,大小約為50~100 nm,分散性較好;由圖8可以看出,所制得的FePO4為片狀,大小約為100~300 nm,分散性較好。

圖7 FePO4·2 H2O樣品的FESEM照片

圖8 FePO4樣品的FESEM照片
2.2.3 FePO4樣品的X-射線衍射分析測定
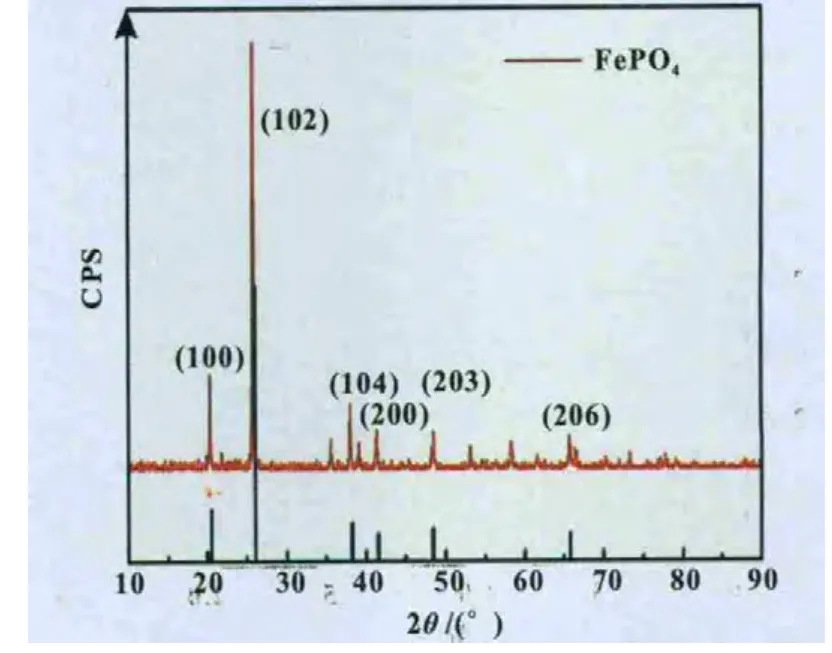
圖9 FePO4樣品及標準卡片29-0715的XRD譜圖
圖9為FePO4樣品(煅燒后)XRD圖譜,從圖譜可見樣品為α-石英晶型(即六方晶型)結構,樣品圖譜特征峰形尖銳,結晶度好,樣品的圖譜和衍射數據與FePO4(卡號:29-0715)標準譜和衍射數據非常相似,且X射線衍射譜中并未觀察到其它的雜質峰,說明通過本實驗制備得到的樣品是純相的FePO4,屬于六方晶系,空間群是P321(150)。說明煅燒后無定形態的FePO4·2 H2O轉變為六方晶系結構的FePO4。
通過對樣品圖譜的指標化計算處理,發現它的晶胞參數與標準PDF卡片29-0715極其相近,結果見表1。
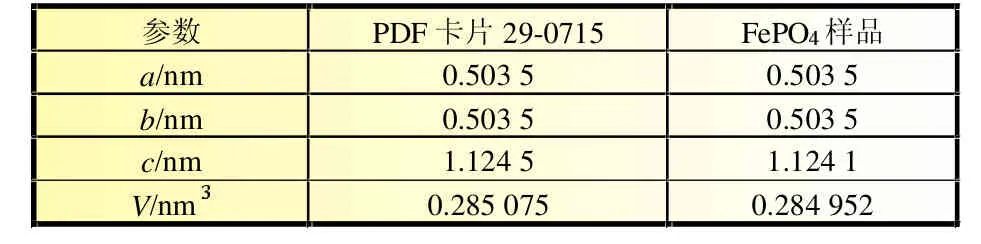
表1 標準PDF卡片29-0715與FePO樣品的晶胞參數
3 結論
本實驗對傳統均相沉淀法制備FePO4粉體工藝進行改進,在低溫醇水體系中,以FeSO4·7H2O和H3PO4為原料,制備FePO4粉體;討論了醇水比例、反應物濃度、攪拌速度和反應時間等因素對FePO4粉體粒度的影響。
研究表明:粉體粒度隨著醇水比例增大而降低;隨著攪拌速度和反應物濃度增大都呈現出先增加后減小的趨勢;反應時間增加使得FePO4粉體粒度逐漸增大。在醇水比例為1∶1,反應物濃度為0.2mol/L,攪拌速度為3 000 r/m in,反應時間為0.5 h,反應溫度為35℃條件下制得的樣品,表征結果顯示其為粒度約50~500 nm的六方晶系結構高純FePO4粉體。在低溫醇水體系中,采用均相沉淀法制備FePO4粉體的工藝路線,具有產物純度高、晶型完整、粒度小、反應溫度低、反應時間和陳化時間短等優勢。
[1]SYLVAIN F,FREDERIC L C,CAROLE B,et al.Comparison between different LiFePO4synthesis routes and their influence on it sphysico chem ical properties[J].Journal of Power Sources,2003,119:252-257.
[2]PROSINIPP,CAREWSKA M,SCACCIA S,et al.Long-term cyclability of nanostructured LiFePO4[J].Electrochem Acta,2003,48(28):4205-4211.
[3]WANGY G,WANGY R,HOSONO E,etal.The design of a LiFePO4/carbon nanocomposite w ith a core-shell structure and its synthesis by an in situ polymerization restrictionmethod[J].Angew Chem Int Ed,2008,47(39):7461-7465.
[4]高飛,唐致遠,薛建軍,等.噴霧干燥-高溫固相法制備納米LiFe-PO4與LiFePO4/C材料及性能研究[J].無機化學學報,2007,23(9):1603-1608.
[5]MOON Y,PARK H K,KIMD K,eta1.Preparation ofmonodisperse and spherical zirconia powders by heating of alcohol-aqueous salt solution[J].JAm Ceram Soc,1995,78(10):2690-2694.
[6]FANG C S,CHEN Y W.Preparation of titania particles by thermal hydrolysis of TiCI4in propanol solution[J].Preparation of titania particles by thermal hydrolysis of TiCI4in propanol solution[J].Materials Chemistry and Physics,2003,78(3):739-745.
[7]GOSTA A O.Dielectric constants of some organic solvent water m ixtures at various temperatures[J].Journal of the American Chemical Society,1932,54(11):4125-4139.
[8]陳建清.超細二氧化鈰制備及其化學機械拋光機理研究[D].鎮江:江蘇大學,2004.
[9]葉鐵林.化工結晶過程原理及應用[M].北京:北京工業大學出版社,2006:27-60.
[10]易均輝.FePO4和LiFePO4的制備、表征及電化學性能研究[D].廣西:廣西大學,2010.
