基態Lin(n=2,3)結構與完全解析勢能函數
2014-06-06 01:00:32路俊哲祝恒江
原子與分子物理學報 2014年3期
關鍵詞:優化
路俊哲,祝恒江
(新疆師范大學物理與電子工程學院,烏魯木齊830054)
1 引 言
鋰是自然界中最輕的金屬,呈銀白色,是唯一能在常溫與氮氣反應的堿金屬元素,具有非常活潑的化學性質.在冶金工業上,利用鋰能與氧、氮、硫等物質發生強烈反應的性質來充當除氣劑和脫硫劑,能有效的清理雜質.鋰又稱“金屬味精”,將其它一些金屬中加入少量鋰,能大大改善原來金屬的性能,如鋰鎂合金、鋁鋰合金、鋰鈹合金等化合物具有質量輕、抗拉強度大、抗腐蝕性強,加工性能好等特點,被廣泛應用于衛星、載人宇宙飛船、飛機等結構材料.鋰又被譽為“能源金屬”,若將鋰或鋰的化合物制成高能燃料,用于火箭、潛艇或飛機的推進劑,具有燃燒溫度高、燃速大、比沖量極高等優點;鋰電池以其壽命高、比能量高、放電平衡、工作溫度范圍寬等特點而廣泛應用于各種領域,是新型的環保節能電池.鋰的同位素6Li具有較強的捕捉低速中子的能力,可以用來控制鈾反應堆中核反應發生的速度,同時還可用于將來核動力飛機和宇宙飛船等,是原子能工業中的“高能金屬”.因此,鋰被視為元素周期表中最令人感興趣的化學元素之一.
多年以來,鋰材料的實驗和理論研究都十分活躍,主要集中在鋰合金材料、鋰電池材料及鋰的電學性質等方面.實驗方面,Bréchignac[1]等人通過蒸汽實驗方法研究了鋰團簇的離解途徑和結合能.Dugourd[2]等人通過光致電離技術獲得鋰團簇的電離勢.Vezin[3]等人通過光致電離技術獲得氫化鋰團簇的電離勢.理論方面的報道也很多,其中Beckmann[4]等人利用多種方法研究了Li4和Li6團簇的構成.Jellinek[5]等人利用分子動力學Hartree-Fock自洽場從頭計算的方法研究了Li8團簇的結構及動力學性質.Gardet[6-7]等人采用密度泛函方法研究了小鋰團簇Lin(n≤20)的電子性質和幾何結構.Fournier[8]等人運用局域自旋密度和梯度修正能量泛函理論研究了小鋰團簇Lin(n=5~20)的幾何結構.另外,對Li6,Li4,Li5,和Li2的結構和穩定性也都有相關報道[9-11].
近幾年,富鋰材料的實驗和理論研究主要集中在鋰離子電池、鋰合金等鋰化物.鋰離子電池具有質量輕、發電高壓高、循環能力好、熱穩定性高、綠色環保等特點[12].鋁鋰合金[13-14]是一種密度低、比強度高、彈性模量高的合金材料,被廣泛應用于航空、航天等領域.LiB合金[15]作為陽極材料所裝配的單元電池在最高電壓和放電時間兩方面具有更好的性能.LiOH[16-17]具有較強腐蝕性,被廣泛應用于化學化工、玻璃、陶瓷等行業,同時還是生產鋰基潤滑脂、堿性蓄電池電解液的主要原料之一,在汽車工業和冶金機械工業中具有良好的應用前景.Li3N具有較強的吸放氫性能,已經成為新型高容量儲氫材料,其結構與性質得到廣泛關注[18-19].
鋰和富鋰材料的理論和實驗研究都十分活躍,但關于Li2,Li3分子及其離子的勢能函數研究卻少有報道.據作者所知,目前有耿振鐸[20-21]等人利用密度泛函方法(B3LYP)和二次組態相互作用方法(QCISD)給出了Li2基態分子結構及勢能函數;施德恒[22]等人使用SAC方法對基態Li2分子進行了平衡幾何及諧振頻率的優化計算,且擬合得到了五參量 Murrell-Sorbie勢能函數;楊建會等人[11,23]利用芶清泉教授提出的改進的排列通道量子力學方法(MACQM)預測Li3分子具有正三角形的構型.考慮到上述研究的分子結構描述不完整或結論與實驗值有較大偏差,特別是關于Li3分子基態勢能函數研究卻未見報道.鑒于此,Li2及Li3分子及其離子的勢能函數值得進一步進行理論研究.本文使用Gaussian03程序包,采用單雙取代耦合簇(CCSD)方法,選擇基組6-311+g(2df),對Li2分子的基態進行優化計算,采用十一參量 Murrell-Sorbie函數,運用最小二乘法擬合得到Li2分子基態解析勢能函數,給出與實驗值符合很好的光譜常數.使用同樣的方法和基組,對Li3分子的基態結構進行優化計算,采用多體項展式法,考慮線性系數和非線性系數之間的關聯,得到Li3分子基態完全解析勢能函數.勢能面靜態特征表明,該勢能函數再現了Li3分子基態全部平衡結構特征.
2 理論計算
2.1 Li2基態分子結構及勢能函數
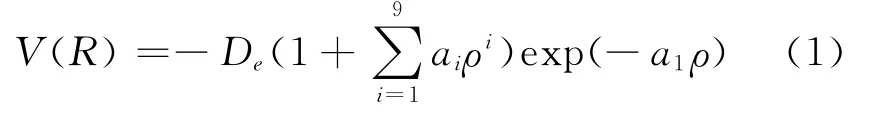
Li2分子是一個簡單體系,但事實表明對Li2分子解析勢能函數的理論研究卻相當困難.原因在于,理論計算時,同一方法不同的基組,甚至不同的初始猜測,都會使計算結果有較大差異.對采用單點能掃描擬合方法得到雙原子分子勢能函數而言,選取一個合理的勢能函數關系十分重要,且單點能的掃描方式和初始猜測的確定也非常關鍵.Murrell-Sorbie函數能準確反應基態雙原子分子的排斥支和吸引支,正確反應其離解極限和長程勢,是用于描述雙原子分子勢能函數的最佳選擇.為提高勢能函數的精度,采用十一參量Murrell-Sorbie函數,其形式為:式中ρ=R-Re,R為核間距,Re為平衡間距,離解能De及a1-a9為 Murrell-Sorbie函數參數.為了確定(1)式,需確定Re、De、a1-a9.首先,采用單雙取代耦合簇(CCSD)方法,參考Re、De、諧振頻率ωe及電子態的實驗數據及已有的理論計算結果,選用不同基組對Li2分子進行優化計算,確定最優基組為6-311+g(2df),計算得到相應數據和結果列表1.然后,采用CCSD方法,選擇6-311+g(2df)基組,對Li2分子進行單點能掃描,將得到的一系列單點勢能值采用最小二乘法進行擬合,得到Murrell-Sorbie函數參數De和a1-a9列表2.由(1)式可得擬合參數與力常數的關系:
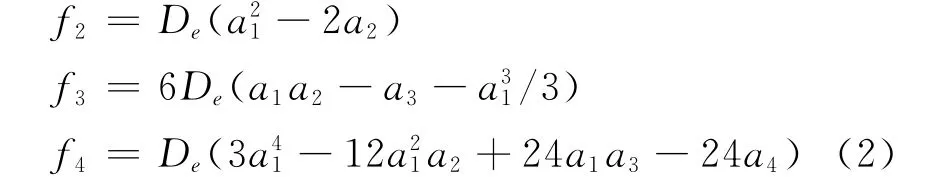
其中力常數f2、f3、f4與光譜常數的關系為

其中μ為單個原子的約化質量,ωe為諧振頻率,xe為非諧振動因子,c為真空光速,Be和αe為剛性和非剛性轉動因子,Drot為離心畸變常數.由(2)、(3)兩式和表2,得到與實驗值符合很好的Li2分子光譜常數列于表3.
從表1、表2和表3可以看出,與實驗值和其他理論方法所得的理論結果相比較,本文所得結果與實驗值符合很好.另外,與優化計算值相比,擬合得到的De和ωe與實驗值符合的更好,且相應數值均好于文獻值.說明擬合得到的勢能函數能夠正確描述Li2分子基態勢能函數性質.圖1給出Li2分子的基態勢能曲線,從中可以看出:在計算范圍內單點能掃描得到的能量點與擬合曲線完全重合(其中實線為擬合結果,圓圈為單點能掃描結果).
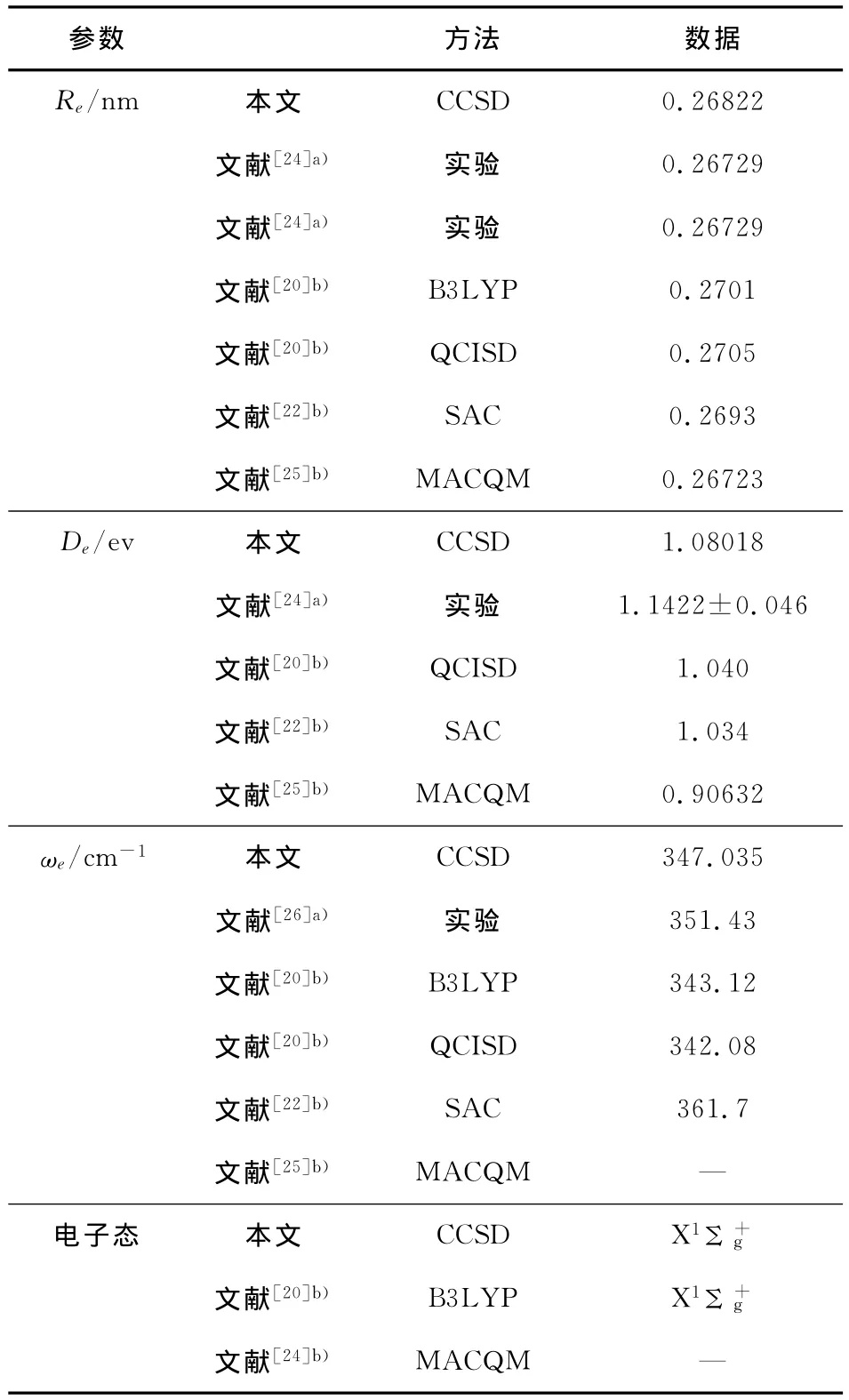
表1 Li2分子的結構參數Table 1 The optimized structural parameters for Li2
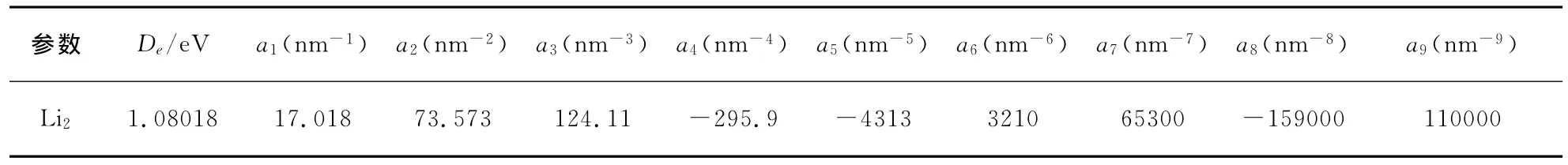
表2 Li2分子離解能、Murrell-Sorbie函數參數Table 2 The dissociation energy and M-S potential energy function parameters for Li2

表3 Li2分子的光譜常數Table 3 The spectroscopic constants for Li2
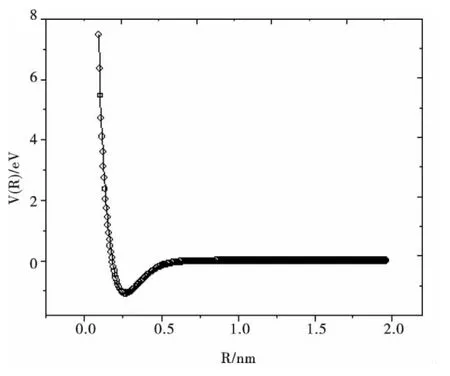
圖1 Li2分子的勢能曲線Fig.1 The potential energy curve of Li2
2.2 Li3分子的平衡結構及力學常數
Li3分子與Li2分子的情況不同,Li2分子僅有D∞h一種穩定構型,其電子態為X1Σ+g.而Li3分子則可能有C2v,C∞v,D∞h,D3h等四種穩定構型.本文采用CCSD方法,選擇6-311+g(2df)基組,優化計算Li3分子二重態,得到C2v,C∞v,D∞h,三種穩定構型,其基態平衡構型為C2v,電子態為2B2.Li3分子三種穩定構型的結構參數和離解能列于表4,C2v構型的振動頻率與力常數列于表5.

表4 Li3分子基態線型、角型構型參數和離解能Table 4 The structure parameters and the dissociation energies of line-and angle-type Li3

表5 Li3分子構型振動頻率與力常數Table 5 The structure parameters of ground state and the force constants for Li3
2.3 基態Li3分子的離解極限
在多原子分子勢能函數的研究中,正確的離解極限是必不可少的.因為在表述原子,分子及其離子的勢能函數中,必須知道在分子離解極限中各原子和原子團的電子狀態,才能給出多體展式中各級展式的解析勢能函數,從而得出多原子分子的勢能函數表達式.與雙原子的情況一樣,要確定多原子分子正確的離解極限,首先要確定電子態,利用從頭計算結合電子組態法的分析可以確定電子態,由分子反應靜力學原理可確定分子可能的電子態和離解極限.計算結果表明Li3分子的基態平衡構型為C2v,電子態為2B2.因此Li3分子可按分離原子法指出的方式離解,又2Sg為Li原子的基電子態,X2B2為Li3分子的基電子態,符合能量最優原理,結合原子分子反應靜力學中的微觀過程傳遞性原理和Li2(X1Σ+g)分子的離解極限,故確定Li3分子的離解極限為:
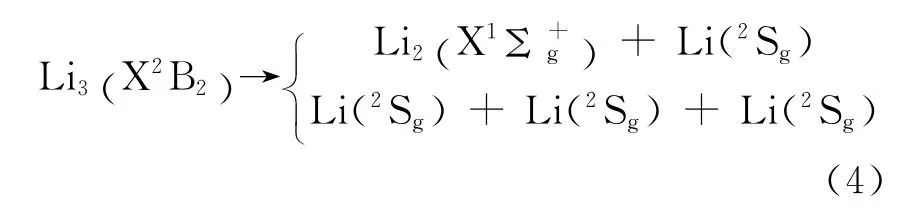
2.4 Li3分子基態完全解析勢能函數
根據多體項展式理論,如果設基態原子的能量為零,則基態B3分子的多體項展式勢能函數可寫為:
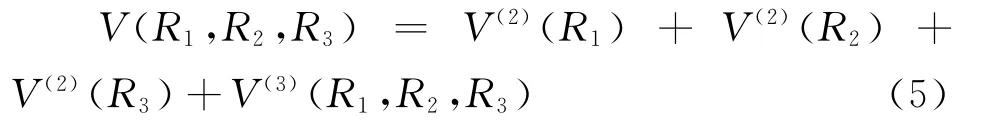
(5)式中R1=R12,R2=R23,R3=R31;V(2)(Ri)(i=1,2,3)為兩體項,可由(1)式得到;V(3)(Ri)(i=1,2,3)為三體項,其形式為:
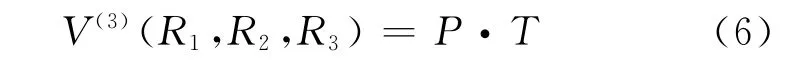
其中P為多項式,T為量程函數,分別為:
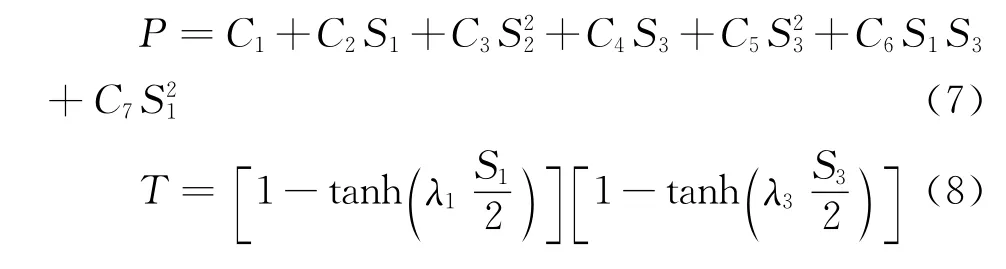
Li3分子的基態構型為C2v.為簡化多體項形式,(6)、(7)兩式是以C2v構型為參考構型,并采用對稱內坐Si(i=1,2,3),內坐標ρi向對稱性坐標變換如下:
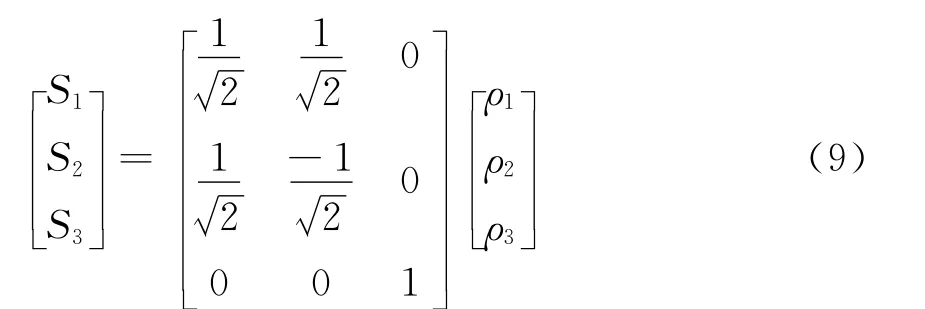
其中內坐標ρi=Ri-R0i(i=1,2,3),優化內坐標中的S2對R1和R2的交換是反對稱的,但R1和R2交換之后分子結構是等同的,為了滿足這一物理性質,(6)式中S2只能含有偶次項.(6)、(7)兩式中含有7個線性系數Ci(i=1,2,3,4,5,6,7)和兩個非線性系數λ1、λ3.利用Li3分子平衡態C2v的幾何結構參數、力常數和離解能,以及Ci(i=1,2,3,4,5,6,7)與λ1、λ3的函數關系,對Li3分子勢能面進行非線性優化得到λ1、λ3,進而得到7個線性系數Ci(i=1,2,3,4,5,6,7),結果列于表6.
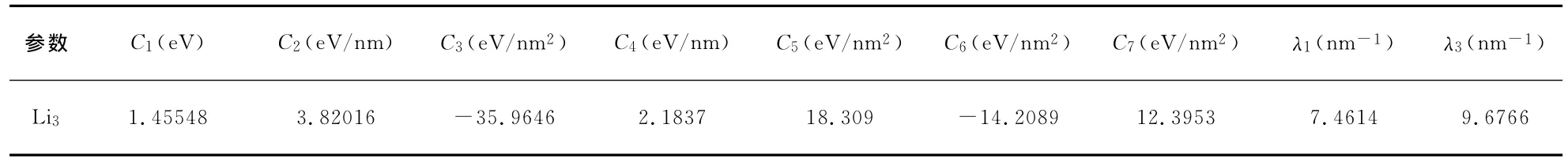
表6 Li3分子勢能函數三體項參數Table 6 Coefficients of 3-body term of potential energy function for Li3
為了直觀分析勢能函數特性,展示對稱性與分子結構以及分子的形成與離解的機理,根據已得出的數據參數,由(4)式圖繪出Li3分子的伸縮振動和旋轉等值勢能圖2~圖4,正確復現了Li3分子的穩態能量和平衡結構.
圖2是固定θ123=72.64981°時,改變鍵長R12,R23的Li3分子勢能函數的伸縮振動等值勢能圖.圖2表明在R12=R23=0.27629nm處有一個極小值1.5964eV,準確再現了Li3分子具有穩定的C2v結構特征,這與優化計算結果完全一致.
圖3將R12=0.27629nm固定在X軸上,以R12鍵的中點為原點建立Y軸,另一Li原子繞R12鍵旋轉時形成的等值勢能圖.從圖3可以看出,當另一Li原子旋轉到θ123=72.64981°,θ231=θ312=53.67509°處,有一個極小值1.5555eV,基本再現了Li3分子穩定的C2v構型;當另一Li原子旋轉到θ321=180°處,有一個極小值1.6610eV,表明Li3分子具有一個穩定的C∞v構型;當另一Li原子旋轉到θ213=180°處,有一個極小值1.6644eV,表明Li3分子具有一個穩定的D∞h構型.由此可知基態Li3分子具有C2v,C∞v和D∞h三個穩態構型.
圖4將R13=0.32733nm固定在X軸上,以R13鍵的中點為原點建立Y軸,另一Li原子繞R13鍵旋轉時形成的等值勢能圖,同樣表明在θ123=72.64981°,θ231=θ312=53.67509°處,有一個極小值1.5964eV,和圖1相應構型的極小值完全一樣,準確再現了Li3分子穩定的C2v構型;當另一Li原子旋轉到θ213=θ132=180°處,有兩個相同的極小值1.7389eV,再現了Li3分子穩定的D∞h構型.
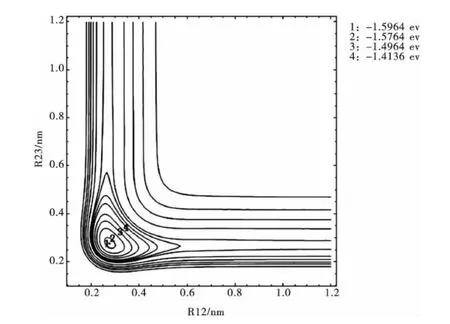
圖2 Li3分子的伸縮振動等值勢能圖Fig.2 Stretched vibrational contour of potential energy for Li3
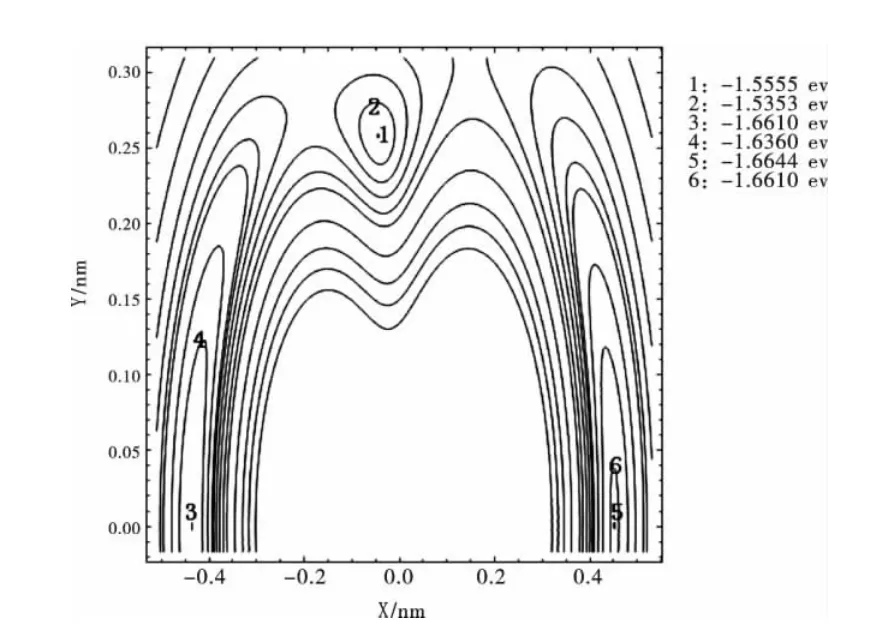
圖3 Li繞Li-Li旋轉的等值勢能圖Fig.3 Rotational contour of potential energy function for Li round Li-Li
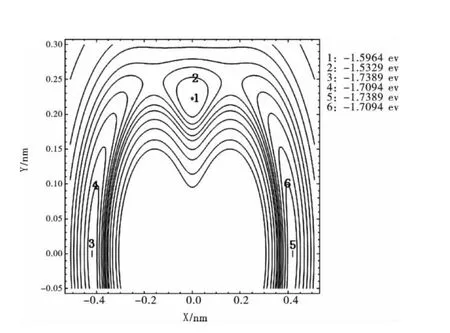
圖4 Li繞Li-Li旋轉的等值勢能圖Fig.4 Rotational contour of potential energy function for Li round Li-Li
圖2~圖4的多體相展式勢能函數結果與優化結果(如表4所示)定性一致,只是優化的結果,C2v構型能量最低,而從多體項展式理論計算的結果看,D∞h構型的能量最低,這是多體項展式理論計算誤差所致.
3 結 論
本文使用Gaussian03程序包,采用單雙取代耦合簇(CCSD)方法,選擇基組6-311+g(2df),對Li2分子的基態進行優化計算,采用十一參量Murrell-Sorbie函數,運用最小二乘法擬合得到Li
2分子勢能函數,得到與實驗值符合很好的光譜常數;對Li3分子的基態平衡結構進行優化計算,采用多體項展式法,利用Li3分子平衡結構C2v的幾何參數、力常數和離解能,以及線性系數Ci(i=1,2,3,4,5,6,7)與非線性系數λ1、λ3的函數關系,先在極小點附近對Li3分子勢能面進行非線性優化擬合得到λ1、λ3,進而得到Ci,得到基態Li3分子的完全解析勢能函數,再現了Li3分子基態全部平衡結構特征,實現了用一個勢能函數同時描述多個極小點的性質或由一種穩定構型的數據確定其它穩定構型的勢能面.
[1]Bréchignac C,Busch H,Cahuzac P,etal.Dissociation pathways and binding energies of lithium clusters from evaporation experiments[J].J.Chem.Phys.,1994,101(8):6992.
[2]Dugourd P,Rayane D,Labastie P,etal.Measurements of lithium cluster ionization potentials [J].Chem.Phys.Lett.,1992,197(4):433.
[3]Vezin B,Dugourd Ph,Rayane D,etal.Ionization potenital measurements of hydrogenated lithium clusters[J].Chem.Phys.Lett.,1993,206:521.
[4]Beckmann H O,Koutecky J.Ab initio SCF and CEPA investigations of stable lithium clusters [J].Chem.Phys.Lett.,1979,67:119.
[5]Jellinek J,Bonaci'cKouteckyV,Fantucci P,etal.Ab initio Hatree-Fock selfconsistentfield molecular dynamicd study of structure and dynamics of Li8[J].J.Chem.Phys.,1994,101(11):10092.
[6]Gardet G,Rogemond F,Chermette H.Density functional theory study of some structural and energetic properties of small lithium clusters [J].J.Chem.Phys.,1996,105(22):9933.
[7]Jones R O,Lichtenstein A I,Hutter J.Density functional study of structure and bonding in lithium clusters Linand their oxides LinO [J].J.Chem.Phys.,1997,106(11):4566.[8]Fournier R,Yi Cheng J B,and Wong A.Theoretical study of the structure of lithium clusters[J].J.Chem.Phys.,2003,119(18):9444.
[9]Plavsi'cD,KouteckyJ,Pacchionl G,etal.Structure and stability of Li4and Li6clusters [J].J.Phys.Chem.A.,1983,87:1096.
[10]Blanc J,Bonaˇci'cKouteckyV,Broyer M,etal.E-volution of the electronic structure of lithium clusters between four and eight atoms[J].J.Chem.Phys.,1992,96(3):1793.
[11]Yang Jian-hui J.Li Ping P.Gou Qing-quan.Formation mechanism and binding energy for equilateral triangle structure of Li3cluster[J].Commun.Theor.Phys.,2005,44(3):525.
[12]Llic D,Kilb M,Holl K,etal.Recent progress in rechargeable nickel/metal hydride and lithium-ion miniature rechargeable batteries [J].Journalof PowerSources,1999,80(1):112.
[13]Lavernia E J,Grand N J.Aluminium-lithium alloys[J].JournalofMaterialsScience,1987,22(5):1521.
[14]Lynch S P.Fracture of 8090Al-Li plate I.Shorttransverse fracture toughness[J].MaterialsScienceandEngineering:A,1991,136(30):25.
[15]Zhang H,Wang L J,Luo Y H,etal.Manufacture of lithium-boron alloy and its properties[J].ChineseJournalofRareMetals.2008,32(2):140(in Chinese)[張浩,王力軍,羅遠輝等.鋰硼合金的制備和性能研究[J].稀有金屬.2008,32(2):140]
[16]Sammy C.Honeycutt H.Recovery lithium as LiOH、H2O from aqueous chloride brines cont aining lithium chloride and sodium chlorider[P].U.SPatent:3597340,1971.
[17]Song S T,Deng X C,Sun J Z,etal.The Progress in the study of the application and preparat ion of lithium hydroxide[J].JournalofSaltandChemicalIndustry,2005,13(2):60(in Chinese)[宋士濤,鄧小川,孫健之等.氫氧化鋰的應用與生產方法研究進展 [J].鹽業與化工,2005,13(2):60]
[18]Novak P,Wagner F R.Electronic structure and magnetism of 3dmetal substituted Li3N [J].JournalofMagnetismandMagneticMaterials,2004,272-276:e269.
[19]Chen Y H,Kang L,Zhang C R,etal.Density functional theory study of the structures and properties of(Li3N)n(n=1~5)clusters[J].Acta Phys.Sin.,2008 ,57(07):4174 (in Chinese)[陳玉紅,康龍,張材榮等.(Li3N)n(n=1-5)團簇結構與性質的密度泛函研究 [J].物理學報,2008,57(07):4174]
[20]Geng Z D,Fan X W,Zhang Y S.Structure and potential energy function of the ground state of XY(H,Li,Na)[J].ActaPhys.Sin.,2006,55(05):2175(in Chinese)[耿振鐸,樊曉偉,張巖松.XY(H,Li,Na)分子基態的結構與勢能函數[J].物理學報,2006,55(05):2175]
[21]Geng Z D.Structure and analytic potential energy function for the ground state and low-lying excited states of Li2,Na2and NaLi [J].Journalof HenanNormalUniversity:NaturalScience,2009,37(1):66(in Chinese) [耿振鐸.Li2,Na2和NaLi分子基態及激發態的結構與勢能函數[J].河南師范大學學報:自然科學版,2009,37(1):66]
[22]Shi D H,Xie A D,Zhu Z L,etal.Analy tical potential energy functions for the electronic states X1,A1and B1Πuof Li2molecule[J].J.At.Mol.Phys.,2004,21:622(in Chinese)[施德恒,謝安東,朱遵略,等.Li2分子X1,A1和B1Πu態的勢能函數 [J].原子與分子物理學報,2004,21:622]
[23]Yang J H.Formation mechanism and binding energy for the atom clusters of Lin(n=2-5)[D].SichuanUniversity,2006(in Chinese)[楊建會.Lin(n=2-5)原子團簇的形成機理及結合能計算[D].四川大學碩士學位論文,2006]
[24]Ruette F,Sánchez M,Anez R,etal.Diatomic molecule data for parametric methods.I[J].J.Mol.Stru.:THEOCHEM,2005,729:19.
[25]Yang J H,Li P,Zhang J P,etal.The MACQM theoretical calculation of binding energy of ground state of Li2[J].J.At.Mol.Phys.,2005,22(03):511(in Chinese)[楊建會,李萍,張建平等.Li2基態結合能的MACQM計算 [J].原子與分子物理學報,2005,22(03):511]
[26]David R.Lide L.CRC handbook of chemistry and physics:Spectroscopic constants of diatomic molecules[M/OL].Florida:CRCPress,2007.http://www.hbcpnetbase.com.
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
能源工程(2022年1期)2022-03-29 01:06:28
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
今日農業(2020年16期)2020-12-14 15:04:59
消費導刊(2018年8期)2018-05-25 13:20:08
家庭影院技術(2018年4期)2018-05-09 07:07:41
電子制作(2017年20期)2017-04-26 06:57:45
