無(wú)菌粉末注射劑生產(chǎn)中可見(jiàn)異物檢查的風(fēng)險(xiǎn)評(píng)估及消減措施
2014-10-10 06:13:18吳耀衛(wèi)
機(jī)電信息 2014年8期
吳耀衛(wèi)
(上海新亞藥業(yè)有限公司,上海201203)
0 引言
2010版《中華人民共和國(guó)藥典(二部)》附錄IB注射劑中將注射劑定義為[1]:注射劑系指藥物與適宜的溶劑或分散介質(zhì)制成的供注入體內(nèi)的溶液、乳狀液或混懸液及供臨用前配制或稀釋成溶液或混懸液的粉末或濃溶液的無(wú)菌制劑。其分為注射液、注射用無(wú)菌粉末與注射用濃溶液。同時(shí),藥典明確規(guī)定:除另有規(guī)定外,注射劑應(yīng)進(jìn)行以下相應(yīng)檢查,即裝量、裝量差異、滲透壓摩爾濃度、可見(jiàn)異物、無(wú)菌、細(xì)菌內(nèi)毒素或熱原。
隨著對(duì)藥品質(zhì)量的不斷提高,人們?cè)谧⑸鋭┲袡z出可見(jiàn)異物的幾率越來(lái)越高,實(shí)際生產(chǎn)與質(zhì)量要求之間的矛盾也相應(yīng)凸現(xiàn),這引起了人們的廣泛關(guān)注。尤其在檢測(cè)依據(jù)、控制判斷標(biāo)準(zhǔn)、檢測(cè)方式、生產(chǎn)過(guò)程中的防患措施等方面更是成為熱門話題[2]。然而,無(wú)菌粉末注射劑生產(chǎn)中的可見(jiàn)異物在人、機(jī)、料、法、環(huán)各環(huán)節(jié)均存在,如何加強(qiáng)監(jiān)控是保證制藥質(zhì)量的一大課題。本文將以抗生素類無(wú)菌粉末注射劑為例,探討無(wú)菌粉末注射劑生產(chǎn)中的可見(jiàn)異物檢查問(wèn)題。
1 可見(jiàn)異物的定義與中國(guó)藥典對(duì)其要求
1.1 可見(jiàn)異物的定義
在2010版中國(guó)藥典二部附錄IX H中將可見(jiàn)異物定義為:可見(jiàn)異物系指存在于注射劑、眼用液體制劑中,在規(guī)定條件下目視可以觀測(cè)到的不溶性物質(zhì),其粒徑或長(zhǎng)度通常大于50μm。
1.2 中國(guó)藥典對(duì)可見(jiàn)異物的要求
藥典規(guī)定:不溶性物質(zhì)粒徑或長(zhǎng)度通常≥50μm,即人工目視在規(guī)定的條件下或用自動(dòng)燈檢機(jī)檢查時(shí)粒徑或長(zhǎng)度≥50μm的不溶性物才算可見(jiàn)異物。
1.3 可見(jiàn)異物的結(jié)果判定
藥典規(guī)定:在靜置一定時(shí)間后輕輕地旋轉(zhuǎn)時(shí)均不得檢出煙霧狀微粒柱,且不得檢出金屬屑、玻璃屑、長(zhǎng)度或最大粒徑超過(guò)2 mm的纖維和塊狀物等明顯可見(jiàn)異物。微細(xì)可見(jiàn)異物(如點(diǎn)狀物、2 mm以下的短纖維和塊狀物等)如有檢出,除另有規(guī)定外,應(yīng)分別符合相應(yīng)規(guī)定。
對(duì)注射用無(wú)菌粉末而言,被檢查的5支(瓶)供試品中,均不得檢出明顯可見(jiàn)異物。若檢出微細(xì)可見(jiàn)異物,每支(瓶)供試品中檢出微細(xì)可見(jiàn)異物的數(shù)量應(yīng)符合注射用無(wú)菌粉末微細(xì)可見(jiàn)異物限度(表1)的規(guī)定;若有1支(瓶)不符合規(guī)定,另取10支(瓶)同法復(fù)試,均應(yīng)符合規(guī)定。

表1 注射用無(wú)菌粉末微細(xì)可見(jiàn)異物限度
然而,針對(duì)與粉針劑分裝相關(guān)的無(wú)菌原料藥,藥典又規(guī)定:5份被檢查的供試品中,均不得檢出明顯可見(jiàn)異物。若檢出微細(xì)可見(jiàn)異物,每份供試品中檢出微細(xì)可見(jiàn)異物的數(shù)量應(yīng)符合無(wú)菌原料藥微細(xì)可見(jiàn)異物限度(表2)的規(guī)定;若有1份不符合規(guī)定,另取10份同法復(fù)試,均應(yīng)符合規(guī)定。

表2 無(wú)菌原料藥微細(xì)可見(jiàn)異物限度
2 可見(jiàn)異物種類及產(chǎn)生原因
存在于注射劑中目視可常見(jiàn)的可見(jiàn)異物可以分為下列幾種:金屬類可見(jiàn)異物、玻璃類可見(jiàn)異物、纖維類可見(jiàn)異物和其他可見(jiàn)異物[2]。
以抗生素類無(wú)菌粉末注射劑為例,可見(jiàn)異物分類及產(chǎn)生原因如表3所示。
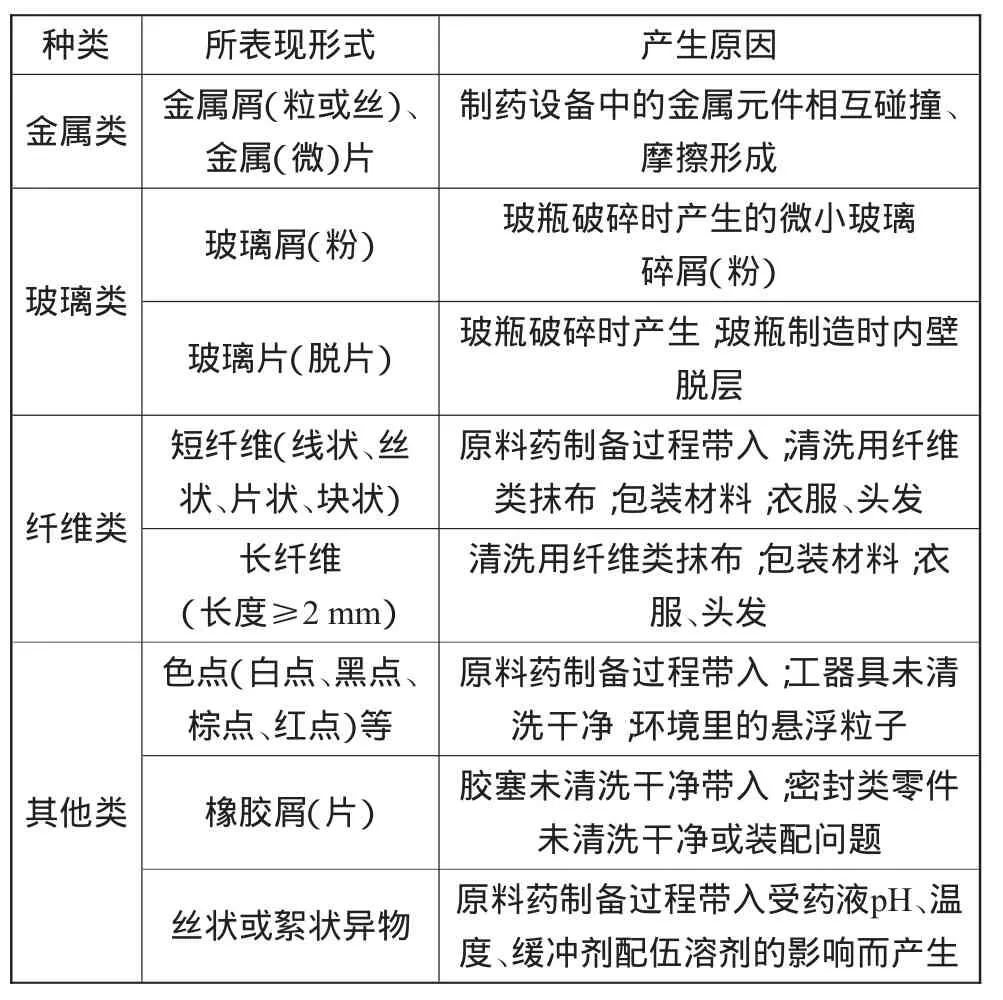
表3 可見(jiàn)異物分類及產(chǎn)生原因
3 可見(jiàn)異物的風(fēng)險(xiǎn)評(píng)估
3.1 從“人、機(jī)、料、法、環(huán)”分析可見(jiàn)異物
以抗生素類無(wú)菌粉末注射劑為例,藥典要求檢查其可見(jiàn)異物是以供試品為切入點(diǎn),而抗生素類無(wú)菌粉末注射劑供試品一般為成品,即經(jīng)分裝、壓塞與軋蓋后,且含瓶子、膠塞、粉體與鋁蓋。因此,分析抗生素類無(wú)菌粉末注射劑可見(jiàn)異物的產(chǎn)生原因可結(jié)合使用魚骨圖分析法,綜合歸納為“人、機(jī)、料、法、環(huán)”5個(gè)方面(表4)。
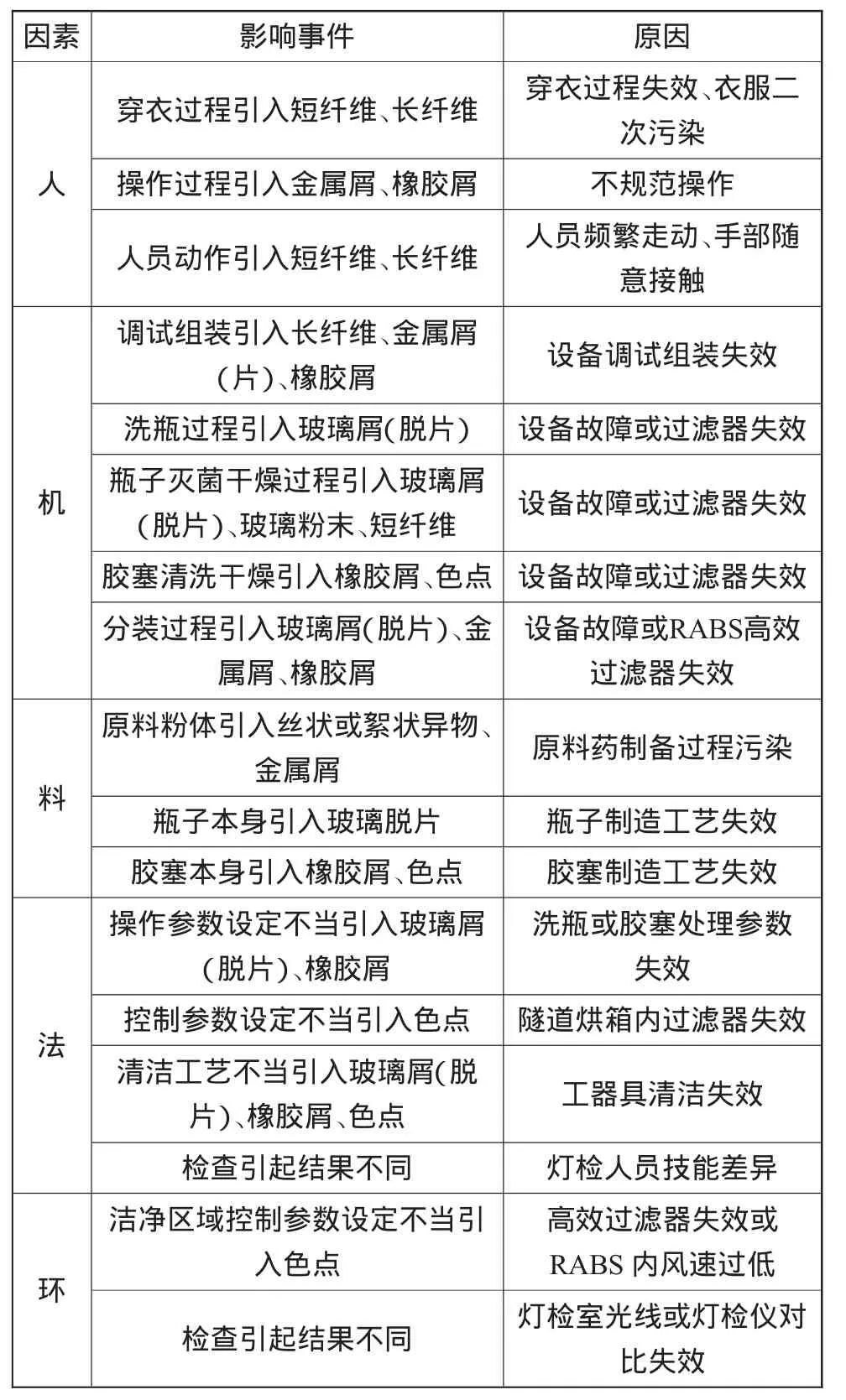
表4 抗生素類無(wú)菌粉末注射劑可見(jiàn)異物的發(fā)生原因
以上統(tǒng)計(jì)是造成抗生素類無(wú)菌粉末注射劑可見(jiàn)異物的發(fā)生原因匯總,現(xiàn)對(duì)其進(jìn)行風(fēng)險(xiǎn)評(píng)估。
3.2 用FMEA法對(duì)可見(jiàn)異物進(jìn)行風(fēng)險(xiǎn)評(píng)估
質(zhì)量風(fēng)險(xiǎn)評(píng)估必須依靠相應(yīng)的定性或定量的風(fēng)險(xiǎn)分析工具,常見(jiàn)工具包括失敗模式與效應(yīng)分析(FMEA)、故障樹(shù)(FTA)、危害與可操作性分析(HAZOP)、危害分析和關(guān)鍵環(huán)節(jié)控制點(diǎn)(HACCP)等[3]。其中,F(xiàn)MEA較為常用,下述以FMEA法對(duì)可見(jiàn)異物進(jìn)行風(fēng)險(xiǎn)評(píng)估。
3.2.1 風(fēng)險(xiǎn)評(píng)估方法簡(jiǎn)介
(1)風(fēng)險(xiǎn)事件識(shí)別。本步驟是對(duì)系統(tǒng)功能或子功能可能導(dǎo)致相關(guān)風(fēng)險(xiǎn)的風(fēng)險(xiǎn)事件進(jìn)行識(shí)別,并確定事件所產(chǎn)生的影響。
(2)風(fēng)險(xiǎn)發(fā)生概率的評(píng)判。在確定所有可能發(fā)生的風(fēng)險(xiǎn)事件及其影響后,應(yīng)對(duì)這些風(fēng)險(xiǎn)事件發(fā)生的概率進(jìn)行判斷。概率判斷的標(biāo)準(zhǔn)并無(wú)統(tǒng)一規(guī)定,具體可根據(jù)事件的復(fù)雜程度來(lái)決定,現(xiàn)風(fēng)險(xiǎn)發(fā)生概率(L)的判斷標(biāo)準(zhǔn)如表5所示。

表5 風(fēng)險(xiǎn)發(fā)生概率的判斷標(biāo)準(zhǔn)
(3)風(fēng)險(xiǎn)事件嚴(yán)重性的評(píng)估。風(fēng)險(xiǎn)事件影響的評(píng)估應(yīng)當(dāng)全面,不僅需評(píng)估風(fēng)險(xiǎn)的直接影響,還應(yīng)評(píng)估這些影響對(duì)于企業(yè)的長(zhǎng)期和廣泛影響。嚴(yán)重性判斷的標(biāo)準(zhǔn)并無(wú)統(tǒng)一規(guī)定,現(xiàn)風(fēng)險(xiǎn)事件嚴(yán)重性(S)的判斷標(biāo)準(zhǔn)如表6所示。
(4)風(fēng)險(xiǎn)等級(jí)的判定。風(fēng)險(xiǎn)等級(jí)的判定可以通過(guò)可見(jiàn)異物風(fēng)險(xiǎn)評(píng)估(表7)進(jìn)行。在矩陣中,風(fēng)險(xiǎn)等級(jí)R=發(fā)生概率(L)×事件嚴(yán)重性(S)。
判定:風(fēng)險(xiǎn)等級(jí)R=20~25為巨大風(fēng)險(xiǎn);R=12~16為重大風(fēng)險(xiǎn);R=4~10為一般風(fēng)險(xiǎn);R<4為輕微或可忽略的風(fēng)險(xiǎn)。
3.2.2 可見(jiàn)異物風(fēng)險(xiǎn)評(píng)估
可見(jiàn)異物風(fēng)險(xiǎn)評(píng)估如表7所示。
4 可見(jiàn)異物檢查指標(biāo)的建立與風(fēng)險(xiǎn)消減措施
從表7可以看出,子問(wèn)題2.2、2.4為風(fēng)險(xiǎn)巨大,子問(wèn)題3.3、4.1為風(fēng)險(xiǎn)重大,這幾個(gè)風(fēng)驗(yàn)是與可見(jiàn)異物相關(guān)且需重點(diǎn)考慮的問(wèn)題。然而,這些風(fēng)險(xiǎn)巨大或重大問(wèn)題歸集起來(lái)便是洗瓶與膠塞清洗干燥工序問(wèn)題,如何消減此類風(fēng)險(xiǎn)呢?對(duì)于抗生素類無(wú)菌粉末注射劑生產(chǎn)而言,關(guān)鍵是可見(jiàn)異物檢查指標(biāo)與風(fēng)險(xiǎn)消減措施的建立。
4.1 可見(jiàn)異物檢查指標(biāo)的建立
對(duì)于抗生素類無(wú)菌粉末注射劑的制劑生產(chǎn)而言,藥典要求是檢查供試品,供試品將由抗生素瓶、原料粉末與膠塞等組成,但此類總組成的微細(xì)可見(jiàn)異物限度為≤8個(gè)。然而,其中與粉針劑分裝相關(guān)的無(wú)菌原料藥,藥典又規(guī)定微細(xì)可見(jiàn)異物限度為≤5個(gè)。因此,抗生素類無(wú)菌粉末注射劑生產(chǎn)環(huán)節(jié)的微細(xì)可見(jiàn)異物限度只能≤3個(gè),這對(duì)于制劑生產(chǎn)過(guò)程來(lái)說(shuō)無(wú)疑是一件困難的事。

表6 風(fēng)險(xiǎn)事件嚴(yán)重性的判斷標(biāo)準(zhǔn)
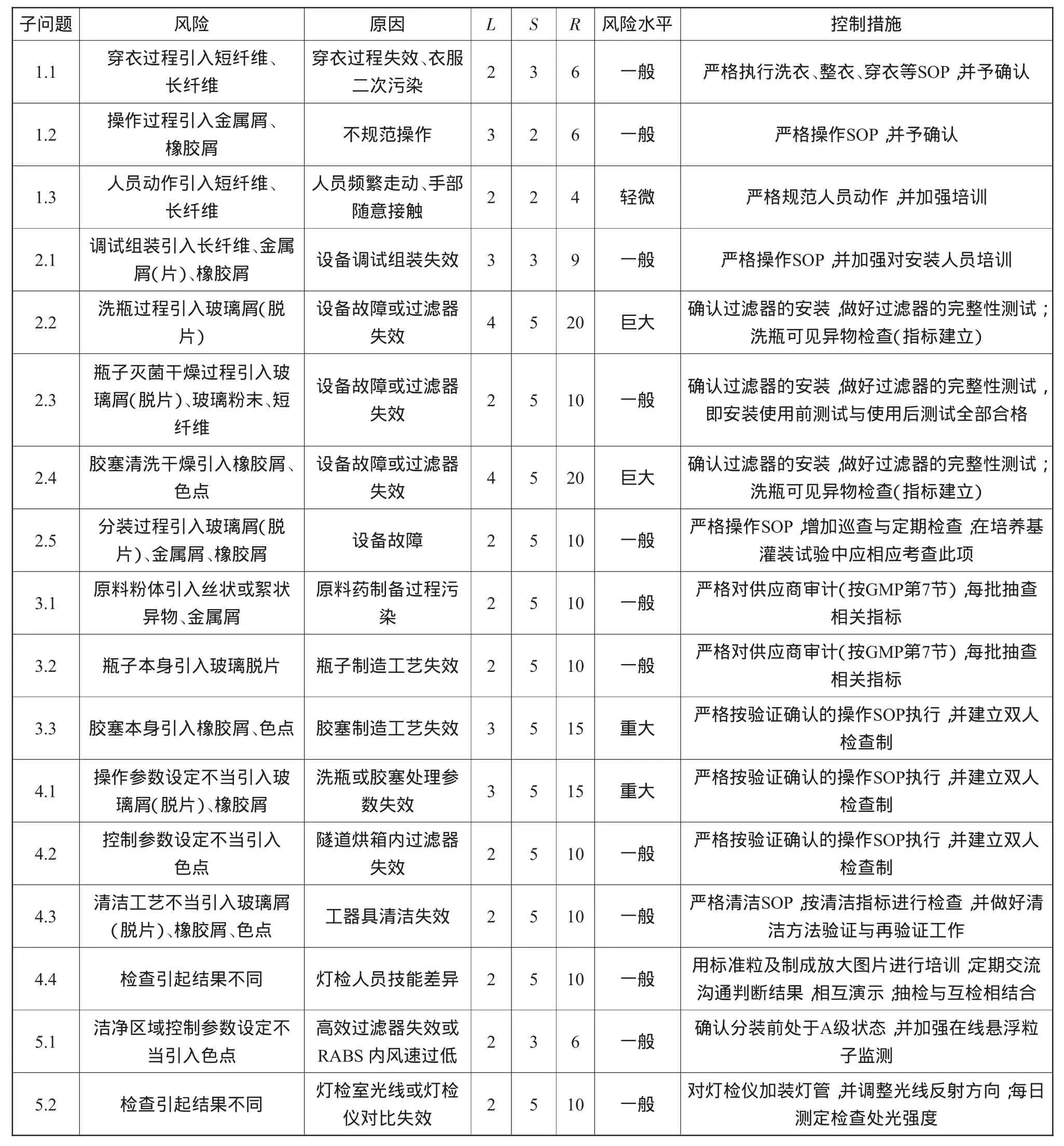
表7 可見(jiàn)異物風(fēng)險(xiǎn)評(píng)估
在尋找建立洗瓶與膠塞清洗干燥工序可見(jiàn)異物檢查企標(biāo)的理念依據(jù)時(shí),我們?cè)谡{(diào)研近3年多個(gè)無(wú)菌原料藥供應(yīng)商產(chǎn)品時(shí)發(fā)現(xiàn),其抽檢可見(jiàn)異物數(shù)量結(jié)果平均為2.33個(gè),且最大可見(jiàn)異物數(shù)量為3個(gè)。為此,對(duì)無(wú)菌原料藥粉末可見(jiàn)異物設(shè)置警戒限和行動(dòng)限,警戒限為≤3個(gè),行動(dòng)限為≤4個(gè)。在此基礎(chǔ)上,洗瓶與膠塞清洗干燥工序可見(jiàn)異物檢查的企標(biāo)就可方便建立,即抗生素瓶清洗后微細(xì)可見(jiàn)異物限度為≤2個(gè),膠塞清洗干燥后微細(xì)可見(jiàn)異物限度為≤2個(gè)。同時(shí),由于現(xiàn)生產(chǎn)均在RABS隔離下生產(chǎn),只有嚴(yán)格控制其余環(huán)節(jié)的微細(xì)可見(jiàn)異物限度,才能使其消減數(shù)量在1個(gè)以下或無(wú),這樣才能使抗生素類無(wú)菌粉末注射劑生產(chǎn)的最終成品達(dá)到藥典要求。
4.2 風(fēng)險(xiǎn)消減措施的建立
4.2.1 嚴(yán)格控制抗生素瓶與膠塞的審計(jì)與抽查
抗生素瓶審計(jì)與抽查要點(diǎn):(1)在玻瓶制造過(guò)程中混入可見(jiàn)異物,如原輔材料、生產(chǎn)環(huán)境中的灰塵、清潔用抹布、破損的玻璃等;(2)在玻瓶質(zhì)量控制過(guò)程中的易產(chǎn)生可見(jiàn)異物,如玻瓶壁上細(xì)微裂紋開(kāi)裂形成的碎屑;(3)在玻瓶原料配方不當(dāng)時(shí)易產(chǎn)生可見(jiàn)異物,如硼硅含量不當(dāng)時(shí)會(huì)引起玻瓶?jī)?nèi)壁脫片。
膠塞審計(jì)與抽查要點(diǎn):(1)選擇與藥物不相溶的橡膠制造膠塞,如鹵化丁基膠、特種橡膠(共聚物);(2)膠塞生產(chǎn)廠房應(yīng)符合GMP要求,在潔凈區(qū)域生產(chǎn)并事先清洗干凈,選用潔凈的輔料與包裝材料(密封包裝),可有效減少橡膠屑的產(chǎn)生;(3)對(duì)于抗生素(頭孢類)產(chǎn)品宜選用四氟乙烯覆膜丁基膠塞,可有效防止異物的產(chǎn)生。
4.2.2 加強(qiáng)檢查人員的培訓(xùn)與考核
可見(jiàn)異物的檢查受人為因素的影響較大,尤其是人工目視燈檢法。其主要因素及消減措施如表8所示[2]。

表8 檢查人員對(duì)可見(jiàn)異物的影響因素及消減措施
4.2.3 與RABS技術(shù)結(jié)合可有效消減
現(xiàn)在設(shè)備均應(yīng)用RABS技術(shù),其能防止可見(jiàn)異物的混入,杜絕環(huán)境和人員對(duì)產(chǎn)品的污染,因?yàn)榭股仡悷o(wú)菌粉末注射劑生產(chǎn)時(shí)微生物的污染和可見(jiàn)異物的混入同時(shí)存在。同時(shí),物料轉(zhuǎn)移的隔離化與自凈區(qū)域的設(shè)置也能有效地消減外界微生物和可見(jiàn)異物對(duì)藥品的污染,確保藥品質(zhì)量的安全性。
4.2.4 文件制度的完善與嚴(yán)格執(zhí)行
(1)建立設(shè)備與相關(guān)零部件清洗的SOP,建立容器具清洗的SOP,保證清洗人員規(guī)范操作。
(2)建立設(shè)備操作的SOP,特別是組裝調(diào)試后的驗(yàn)收要求、過(guò)濾器性能監(jiān)控。
4.2.5 其他消減措施
(1)人員培訓(xùn)。生產(chǎn)人員需經(jīng)過(guò)嚴(yán)格的培訓(xùn)和考核,拿到合格證后方可上崗。
(2)生產(chǎn)安排的強(qiáng)度應(yīng)適中,防止人員因過(guò)度疲勞導(dǎo)致操作不當(dāng)?shù)蕊L(fēng)險(xiǎn)的發(fā)生。
(3)清潔驗(yàn)證及再驗(yàn)證。每個(gè)產(chǎn)品都要做清潔方法的適用性驗(yàn)證,即清潔驗(yàn)證,以所有產(chǎn)品的合格標(biāo)準(zhǔn)中的最低值為驗(yàn)證標(biāo)準(zhǔn)。
5 結(jié)語(yǔ)
本文從可見(jiàn)異物的定義與中國(guó)藥典對(duì)其要求入手,以抗生素類無(wú)菌粉末注射劑為例,分析了可見(jiàn)異物種類及產(chǎn)生原因,繼而對(duì)其進(jìn)行風(fēng)險(xiǎn)評(píng)估,并探討了可見(jiàn)異物檢查指標(biāo)的建立與風(fēng)險(xiǎn)消減的相關(guān)措施。由此可以得出結(jié)論:只有通過(guò)風(fēng)險(xiǎn)評(píng)估,強(qiáng)化有效消減風(fēng)險(xiǎn)措施,特別是相關(guān)工序檢查企標(biāo)的建立,才能使抗生素類無(wú)菌粉末注射劑的可見(jiàn)異物指標(biāo)達(dá)到藥典要求。
[1]國(guó)家藥典委員會(huì).中華人民共和國(guó)藥典(二部)[M].北京:中國(guó)醫(yī)藥科技出版社,2010
[2]周立法,趙小英.芻議注射劑“可見(jiàn)異物檢查”[J].機(jī)電信息,2012(2)
[3]王燕,肖瀟,梁毅.淺析質(zhì)量風(fēng)險(xiǎn)管理在計(jì)算機(jī)化系統(tǒng)驗(yàn)證中的應(yīng)用[J].機(jī)電信息,2011(4)
