低溫循環(huán)色譜法分離H2/HD
2015-02-13 05:04:35翁葵平侯建平任興碧
核化學與放射化學 2015年1期
謝 波,翁葵平,侯建平,李 毅,任興碧,古 梅
中國工程物理研究院 核物理與化學研究所,四川 綿陽 621999
低溫循環(huán)色譜法分離H2/HD
謝 波,翁葵平,侯建平,李 毅,任興碧,古 梅
中國工程物理研究院 核物理與化學研究所,四川 綿陽 621999
低溫循環(huán)色譜法(CCC)是一種有效的氫同位素分離方式。在升級改造后的低溫色譜分離裝置上開展了H2/HD體系的分離研究。結(jié)果表明:原始氘豐度為1.4×10-4的高純氫經(jīng)過CCC 4個流程后,氘豐度達到1.173×10-3;為獲得最佳色譜柱柱效,CCC的進樣量控制在組分峰的容量因子下降10%時較為合適;CCC的雙柱間可互相充當解吸柱與接收柱的角色,在柱結(jié)構(gòu)與程序升溫條件相同的前提下,雙柱間分離效果的差異可能是進樣點的選擇和進樣壓力的不同造成的,與進樣時間無關。
色譜;氫同位素;分離;豐度
氫同位素分離對于聚變堆、聚變-裂變混合堆燃料循環(huán)和氚環(huán)境安全約束十分重要。在聚變?nèi)剂涎h(huán)中,低溫精餾曾經(jīng)被認為是氫同位素分離工藝的最佳方式,因為精餾是十分成熟的工業(yè)技術,在氘的商業(yè)生產(chǎn)中得到實際應用。但這種大規(guī)模的分離工藝,需要操作大量的液化氣體,氫同位素滯留大,這對于氘-氚聚變?nèi)剂涎h(huán)來說是一個缺點,因為聚變?nèi)剂涎h(huán)要求盡可能減小氚在系統(tǒng)中的滯留,氚的稀有、氚的增殖、氚的衰變等特性決定了必須采用一種小滯留的方式進行氫同位素的分離[1]。為建立經(jīng)濟、有效的氫同位素分離系統(tǒng),本課題組自1989年以來,開發(fā)了低溫循環(huán)色譜法(cryogenic cycling chromatography,CCC)這一自制制備色譜技術,先后建立了多套不同處理容量的低溫循環(huán)色譜分離裝置,實現(xiàn)了中試規(guī)模的長期、連續(xù)、安全運行。為了進一步了解分子篩多組分吸附特性、吸附等溫線、氫同位素的傳質(zhì)-擴散-滲透-滯留等色譜分離特征,在科技部ITER專項的支持下,本課題組對低溫色譜分離裝置進行了升級改造,直接瞄準CCC分離工藝在聚變或聚變-裂變?nèi)剂涎h(huán)中得到應用的目標,期望在氫同位素分離實現(xiàn)工程化的目標上前進一步。
1 實驗
1.1 設備與材料
以升級改造后的低溫循環(huán)色譜裝置為實驗平臺(圖1)。高純氫(純度99.999%)、高純氦(純度99.999%),成都金克星氣體公司,高純氫中的氘豐度經(jīng)色譜測量為1.4×10-4(體積分數(shù));5A分子篩,中國科學院大連化學物理研究所;液氮,成都富僑氣體公司;6890N氣相色譜儀,美國安捷倫公司。
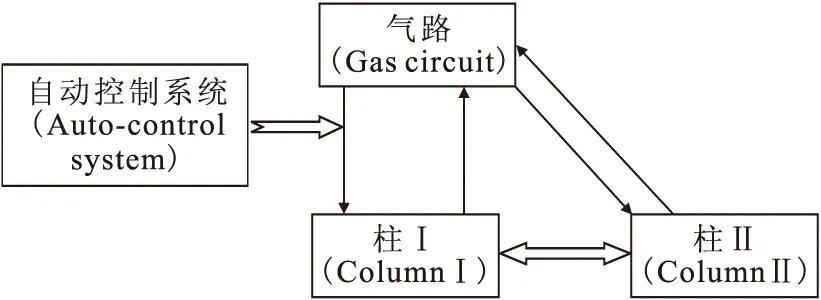
圖1 低溫循環(huán)色譜分離裝置結(jié)構(gòu)簡圖Fig.1 Structural chart of CCC separation device
1.2 實驗流程
實驗流程示于圖2。流程特點、進樣次數(shù)、進樣時間與進樣量列于表1。
2 結(jié)果與討論
2.1 第一次流程
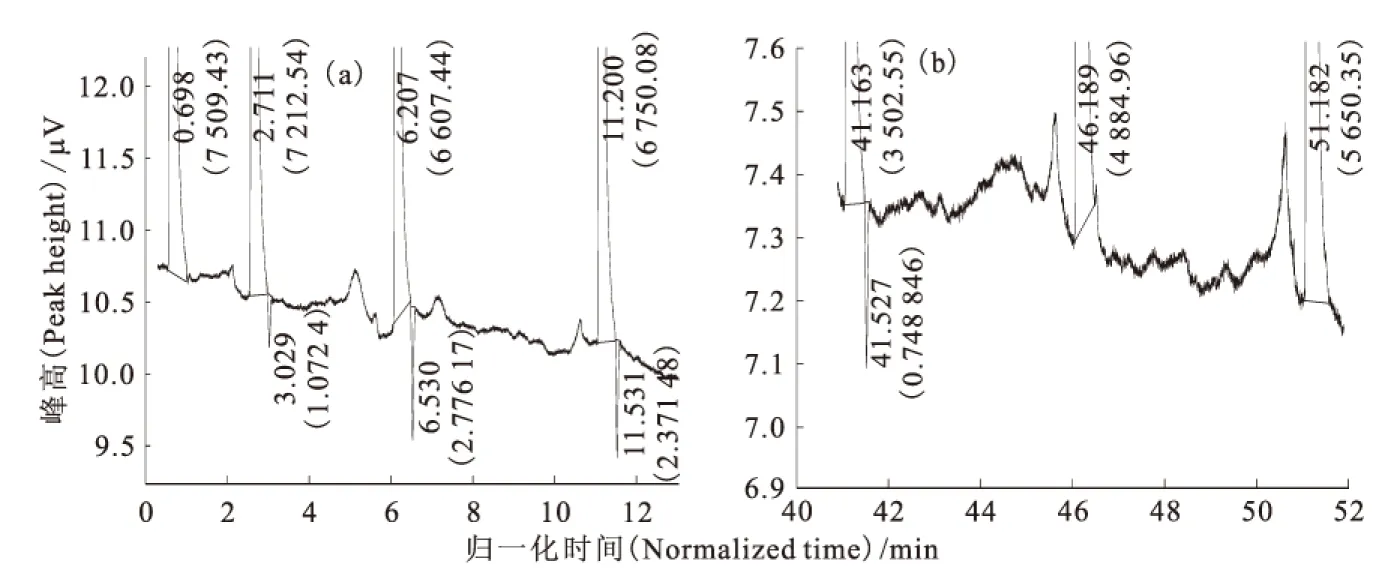
在第一次流程實驗中,色譜柱完成兩次進樣,合計2.19m3高純氫。在氣相色譜儀過程監(jiān)測中,首次出HD負峰和HD正峰的色譜譜圖示于圖3。無論是正峰還是負峰,HD峰一直都在He峰之后。同時,HD負峰呈脈沖趨勢,忽大忽小,幾起幾落,通氦氣加熱歸一化時間為50min時,HD由大負峰逐漸與基線平行,65min時出現(xiàn)明顯的HD正峰,校正后的HD濃度為1.86×10-4(體積分數(shù)),已經(jīng)略高于該氫中的原始氘豐度1.4×10-4(體積分數(shù)),即表明僅僅通過一次流程就明顯出現(xiàn)了氘的濃縮效果。此外,在進樣過程中,柱阻力先降再升,又降后升,出現(xiàn)數(shù)次反復。停止進樣后,柱阻力先升后降,在300kPa左右時趨于平穩(wěn)。這說明進樣過程正常,未發(fā)生串流與倒吸。結(jié)束這一流程時,末尾監(jiān)測無明顯O2和N2峰。以歸一化時間為橫坐標,He峰體積百分比與HD校正濃度(體積分數(shù))為縱坐標作圖,得到第一次流程的運行結(jié)果,示于圖4。從圖4可以看出,由于是初始流程,組分在氣-固相間的吸附分配尚未達到平衡,HD濃度變化無明顯規(guī)律,既有緩慢升高,也有陡降陡升。而He峰百分比是先降后緩慢回升,似乎與HD濃度變化無關。
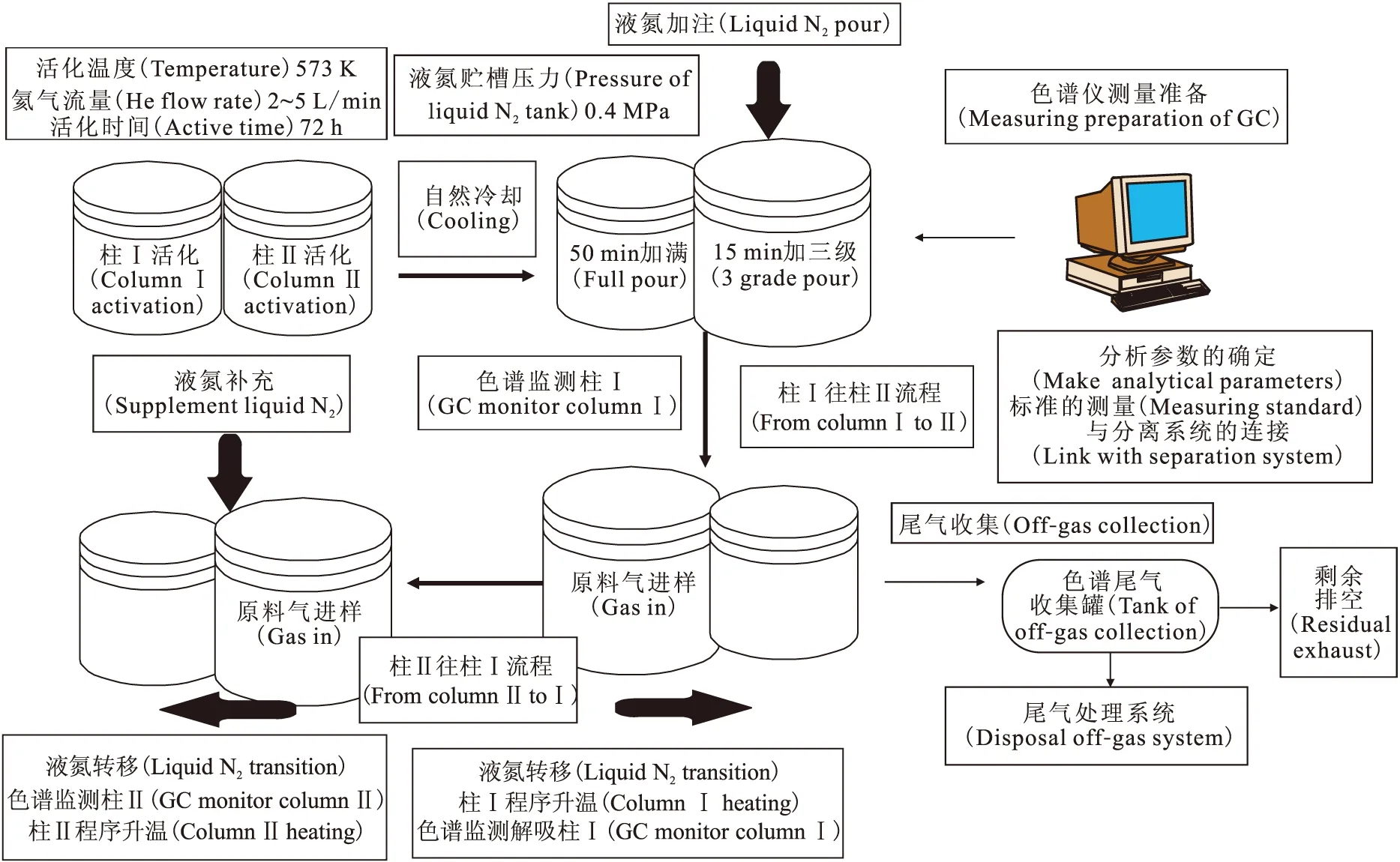
圖2 H2/HD分離實驗流程示意圖Fig.2 Schematic diagram of H2/HD separation flow
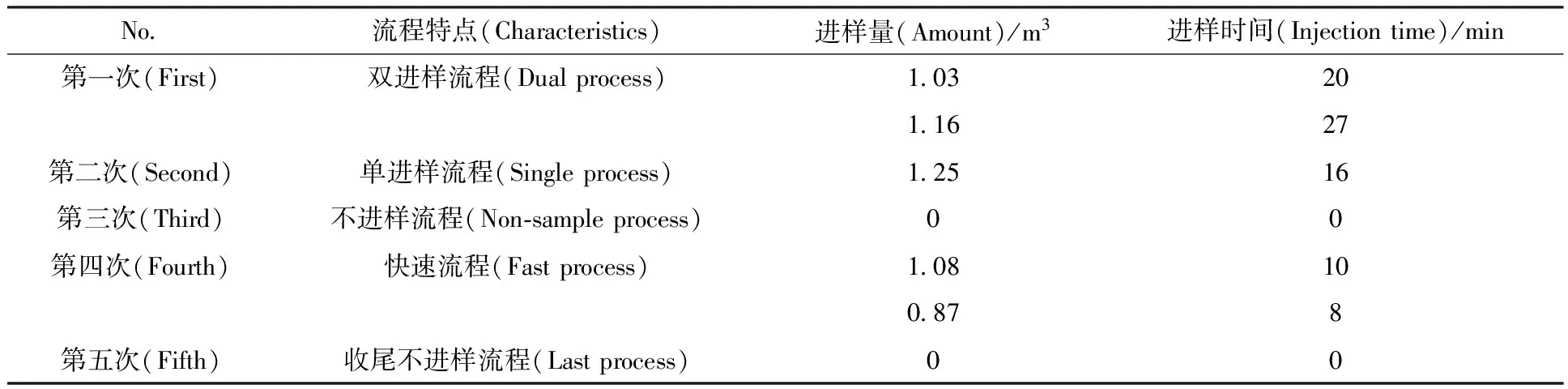
表1 H2/HD分離實驗流程一覽表Table 1 List of H2/HD separation flow
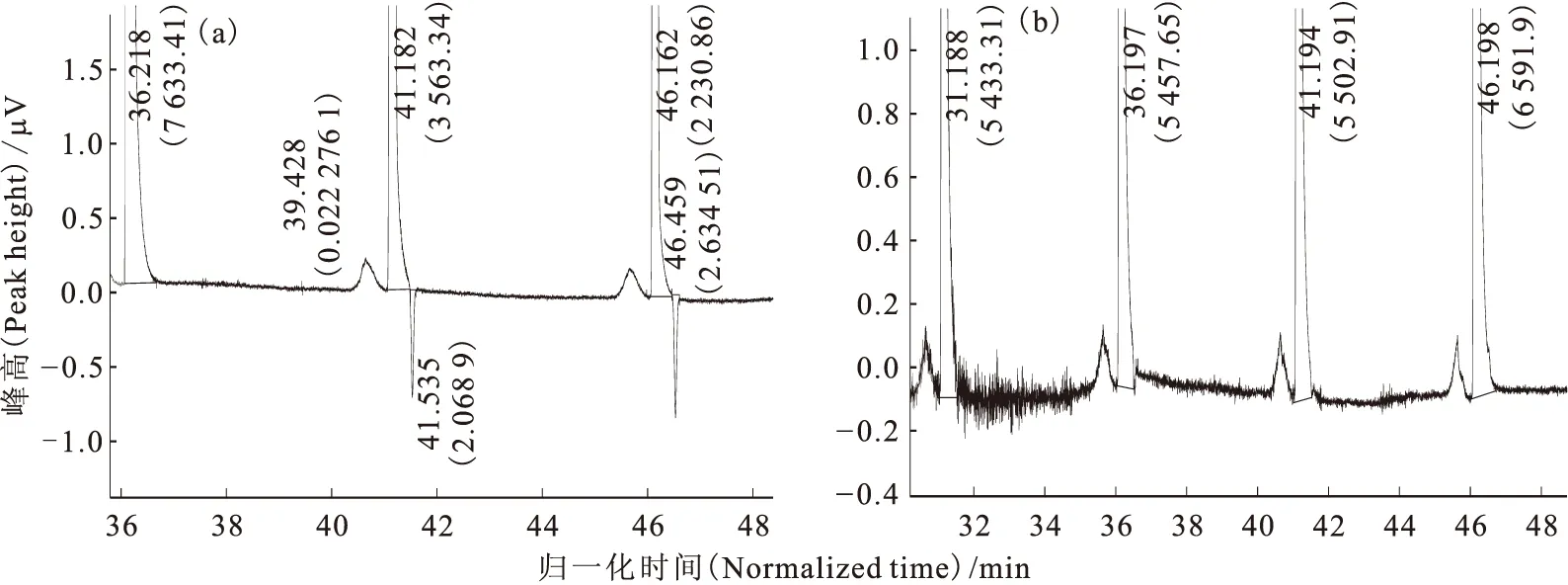
括號中數(shù)值為峰面積(The data in parentheses are peak area)圖3 第一次流程中HD負峰(a)和HD正峰(b)的譜圖Fig.3 Charts of HD negative peak(a)and positive peak(b)in the first flow
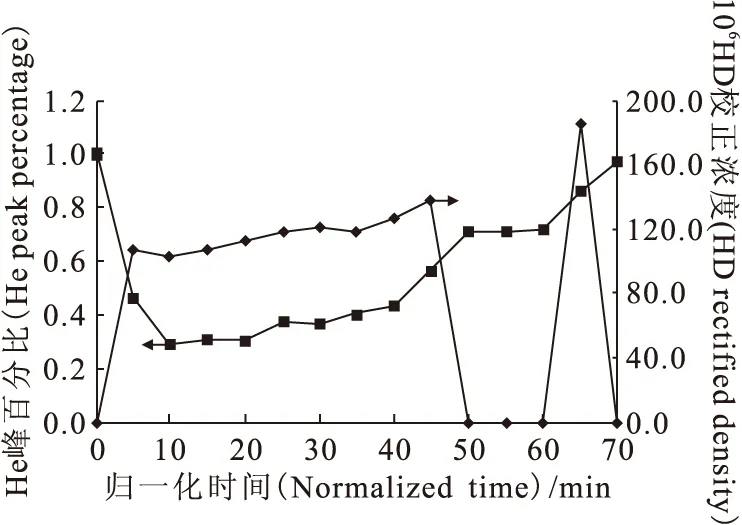
圖4 第一次流程的結(jié)果Fig.4 Results of the first flow
2.2 第二次流程
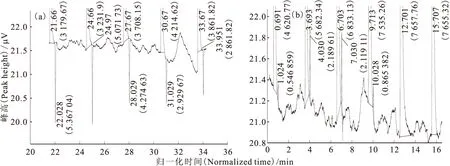
第二次流程中色譜柱完成一次進樣1.25m3高純氫,HD負峰和HD正峰的監(jiān)測譜圖示于圖5。由圖5可以看出,在液氮轉(zhuǎn)移、載氣流量、柱壓力基本保持不變的情況下,與第一次流程相比,第二次流程的結(jié)果既有相似也有差異。相似之處在于貧氘氫均呈脈沖式,忽大忽小;差異之處在于第二次流程中的HD正峰略小,但He峰沒有明顯減小。以歸一化時間為橫坐標,氦峰百分比與HD校正濃度為縱坐標作圖,得到第二次流程的運行結(jié)果,示于圖6。從圖6可以看出,除流程起始點與流程終點外,HD濃度與He峰百分比的變化趨勢是完全一致的,呈現(xiàn)脈沖形式,即He峰百分比下降時HD濃度也下降,He峰百分比上升時HD濃度也上升。這是由于載氣He是以脈沖方式而不是連續(xù)地進入色譜柱,每個脈沖
括號中數(shù)值為峰面積(The data in parentheses are peak area)圖5 第二次流程中HD負峰(a)和HD正峰(b)的譜圖Fig.5 Chart of HD negative peak(a)and positive peak(b)in the second flow
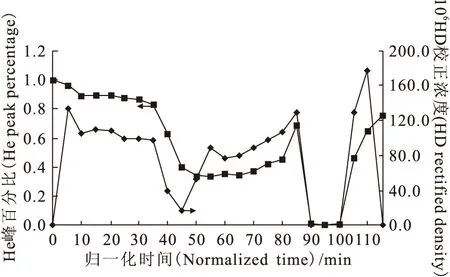
圖6 第二次流程的結(jié)果Fig.6 Results of the second flow
的體積都等于色譜塔板理論中所說的板體積,由于載氣不斷往前沖洗,而固定相分子篩保持不動,組分HD隨載氣移動時被不斷地吸附-解吸,即多次重新分配,顯然第二次流程仍未達到分配的平衡。從圖3、圖5還可以看出,HD負峰位于He峰的后面,He峰對其有拖帶效應,隨著進樣量的增加,HD負峰峰高增加緩慢,但峰寬有所加大。
2.3 第三次流程
第三次流程沒有進樣,HD負峰和HD正峰的譜圖示于圖7。由圖7可以看出,HD正峰面積有較大提高,說明色譜流程在沒有進樣的情況下(去繼續(xù)擠壓前兩次流程產(chǎn)生的濃縮效果),富集濃縮作用更為明顯。反過來看,這可以說明進樣量與色譜柱柱效的關系。進樣量增加時,色譜柱的理論塔板數(shù)N有所下降,這時有兩種情形:一是原料氣適當過量,即在非線性的條件下進行制備性分離,并不會對分離效果產(chǎn)生災難性的影響,這一結(jié)果與加拿大安大略省水電研究室的氣相色譜氫同位素分離演示系統(tǒng)所得到的結(jié)論一致[2];二是原料氣嚴重過量,造成色譜柱柱效類似于指數(shù)下降,使分離能力急劇降低甚至喪失。因此,考慮到制備色譜分離策略中的峰接觸法原理,樣品增加的最高限量將是待收集物(目標物)的色譜峰與最鄰近雜質(zhì)的色譜峰剛好接觸時的進樣量,這個進樣量的大小可利用下式[3]進行估算:
括號中數(shù)值為峰面積(The data in parentheses are peak area)圖7 第三次流程中HD負峰(a)和HD正峰(b)的譜圖Fig.7 Chart of HD negative peak(a)and positive peak(b)in the third flow
(1)
式中:ω為進樣量;ωs為色譜柱的樣品飽和容量;α為分離因子。該式表明,色譜柱的飽和容量越高、分離因子越大,則允許進樣量就越大。色譜柱的飽和容量是指在基本上不降低柱分辨率的情況下所能分離的樣品量,色譜柱容量直接取決于填料的性質(zhì)和容量,還與樣品氣特性及相對分子質(zhì)量和保留值有關。對于色譜柱飽和容量的大小,可根據(jù)以下經(jīng)驗公式[3]進行估算:ωs(mg)≈0.4×柱分離材料的總表面積(m2)
(2)
從此式可以看出,分離材料的表面積越大,色譜柱的樣品飽和容量越高。對于一級分離系統(tǒng)φ32mm×33m而言,5A分子篩的裝填質(zhì)量為14.5kg,則進樣量應該控制在組分峰的容量因子(k)下降10%左右比較合適。以歸一化時間為橫坐標,He峰百分比與HD校正濃度為縱坐標作圖,得到第三次流程的結(jié)果,示于圖8。從圖8可
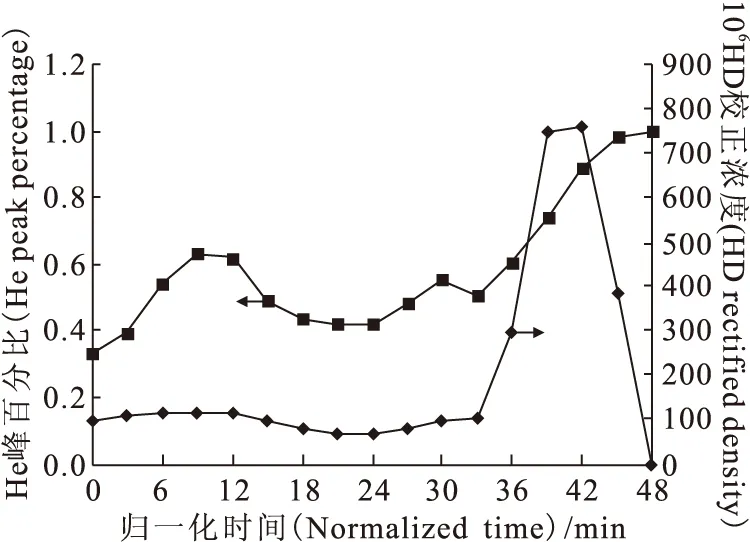
圖8 第三次流程結(jié)果圖Fig.8 Results of the third flow
以看出,HD濃度變化的劇烈程度大大降低,在后期出現(xiàn)了兩個濃度極大點而形成峰形,He峰百分比總體上是逐漸增加,這些現(xiàn)象說明第三次流程出現(xiàn)了分配平衡的特征。
2.4 第四次流程
第四次流程是在短時間內(nèi)完成了兩次進樣,譜圖(圖9)監(jiān)測結(jié)果表明,HD正峰峰形與數(shù)量優(yōu)異,柱Ⅰ與柱Ⅱ互相充當解吸柱與接收柱的角色,在色譜柱結(jié)構(gòu)與程序升溫條件相同的前提下,兩者間的差異可能是進樣點的選擇和進樣壓力的不同造成的,與進樣時間無關。
2.5 第五次流程
經(jīng)過四次流程后,氘已經(jīng)被濃縮了一個量級,其豐度已達到1.173×10-3。第五次流程屬于實驗收尾階段,監(jiān)測譜圖示于圖10。由圖10可以看出,無論是與第三次流程相比,還是與第四次流程相比,第五次流程反映出來的HD正峰峰形更佳,數(shù)據(jù)離散點更少,說明色譜柱分離狀態(tài)趨于穩(wěn)態(tài),分子篩的吸附-解吸過程趨于穩(wěn)定,氫-氘體系的傳質(zhì)性能與動態(tài)分布更加接近于理想狀態(tài)。第五次流程的結(jié)果示于圖11。從圖11可以看出,除個別點以外,He峰百分比基本保持不變,HD濃度出現(xiàn)了兩個峰形,說明第五次流程已經(jīng)是色譜塔板理論所說的分配平衡狀態(tài),吸附分配比為常數(shù),與組分He、HD在塔板上的量無關。
2.6 與過去H-D流程的比較
將五次流程的HD校正濃度與2003年的實驗數(shù)據(jù)[4]放在一起,并將歸一化時間改為時間階段后,結(jié)果示于圖12。從圖12可以看出,雖然不同流程之間存在實驗條件的差異,雙進樣、單進樣、非進樣,但測量的D豐度結(jié)果還是存在一致性或相似的地方。與2003年的數(shù)據(jù)相比,從曲線形狀來講,不考慮時間階段的話,第四次流程和第五次流程與2003年數(shù)據(jù)極為相似,即使是第三次流程,其曲線形狀也與第五次流程的后半部分相似。即在將高純氫中原始D豐度濃縮一個量級的過程中,升級改造后的色譜分離裝置依然有效。
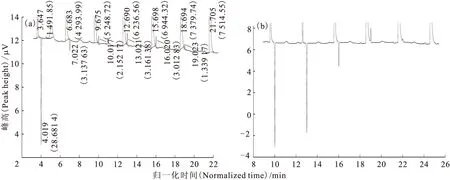
括號中數(shù)值為峰面積(The data in parentheses are peak area)(a)——柱Ⅰ→柱Ⅱ(Column Ⅰ→Column Ⅱ),(b)——柱Ⅱ→柱Ⅰ(Column Ⅱ→Column Ⅰ)圖9 第四次流程中的HD峰譜圖Fig.9 Charts of HD peaks in the fourth flow

括號中數(shù)值為峰面積(The data in parentheses are peak area)(a)——柱Ⅰ→柱Ⅱ(Column Ⅰ→Column Ⅱ),(b)——柱Ⅱ→柱Ⅰ(Column Ⅱ→Column Ⅰ)圖10 第五次流程中的HD峰譜圖Fig.10 Charts of HD peaks in the fifth flow
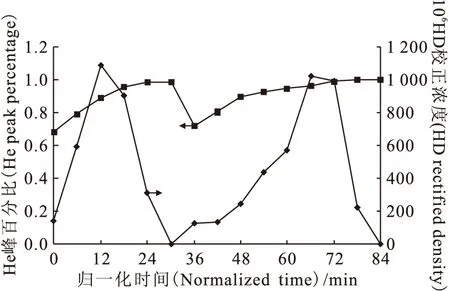
圖11 第五次流程結(jié)果圖Fig.11 Results of the fifth flow
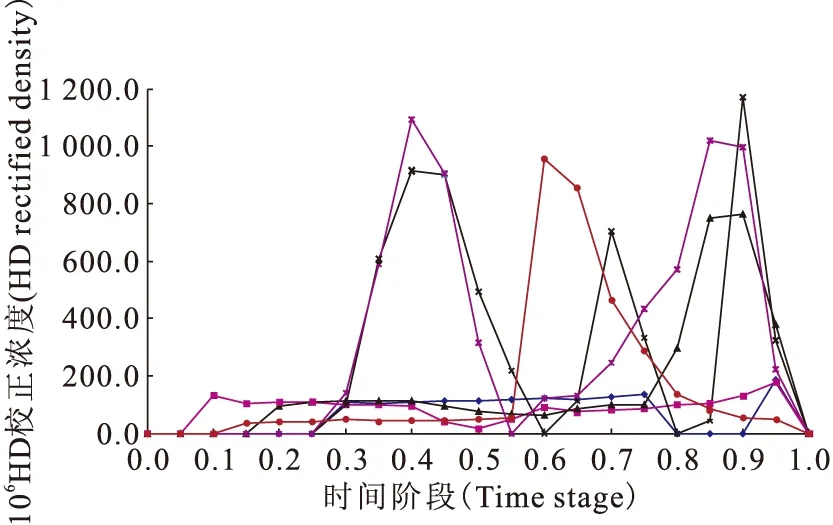
◆——第一次流程(First flow),■——第二次流程(Second flow),▲——第三次流程(Third flow),×——第四次流程(Fourth flow),*——第五次流程(Fifth flow),●——2003年數(shù)據(jù)(2003data)圖12 五次流程結(jié)果與2003年數(shù)據(jù)的比較Fig.12 Comparison between result of five flows and 2003data
3 結(jié) 論
(1)采用低溫循環(huán)色譜的方式,原始豐度的高純氫原料氣經(jīng)過4次流程后,氘豐度被濃縮一個量級,達到1.173×10-3;
(2)對于尺寸較大的低溫循環(huán)色譜系統(tǒng)而言,進樣量應控制在組分峰的容量因子k下降10%時較為合適;
(3)柱Ⅰ與柱Ⅱ可互相充當解吸柱與接收柱的角色,在柱結(jié)構(gòu)與程序升溫條件相同的前提下,兩者間的差異可能是進樣點的選擇和進樣壓力的不同造成的,與進樣時間無關。
[1]Holtkamp N. An overview of the ITER project[J]. Fusion Eng Des,2007,82: 427.
[2]Cheh C H. Large scale gas chromatographic demonstration system for hydrogen isotope separation[J]. Fusion Technol,1988,14: 567-573.
[3]劉虎威.氣相色譜方法及應用[M].北京:化學工業(yè)出版社,2007:32.
[4]謝波,劉云怒,侯建平,等.氣相色譜法濃縮氘的研究[J].核技術,2005,28(12):934-936.
H2/HD Separation by the Cryogenic Cycling Chromatography
XIE Bo,WENG Kui-ping,HOU Jian-ping,LI Yi,REN Xing-bi,GU Mei
Institute of Nuclear Physics and Chemistry,China Academy of Engineering and Physics,Mianyang 621999,China
The cryogenic cycling chromatography(CCC)is an effective way for hydrogen isotopes separation. The separation study of H2/HD system has been performed on the cryogenic chromatographic device after reform and upgrade. It was concluded that the deuterium abundance of pure hydrogen can reach 1.173×10-3by four flows from the original abundance 1.4×10-4. In order to acquire the best chromatographic column efficiency,the injection volume of CCC must be regulated with capacity factor of ingredient peak decreasing 10%. Dual columns of CCC can serve as the desorption column and receipt column each other. Under same conditions of columns structure and temperature programmed,the discrepancy of separation result for dual columns may be caused by the choice of the sample injection point and different sample injection pressures,but not by the sample injection time.
chromatograph; hydrogen isotopes; separation; abundance
2014-03-12;
2014-09-05
科技部ITER計劃專項(2011GB111004)
謝 波(1975—),男,湖北潛江人,碩士,副研究員,主要從事同位素分離與氚工藝研究
O643.14
A
0253-9950(2015)01-0018-06
10.7538/hhx.2015.37.01.0018
