利用改良的CRISPR/Cas9基因編輯系統構建HeLa細胞SND1基因敲除穩定株
2015-03-01 10:10:22劉郁瑩崔曉騰高星杰趙秀娟付雪葛林蘇超楊潔
天津醫科大學學報 2015年6期
劉郁瑩,崔曉騰,高星杰,趙秀娟,付雪,葛林,蘇超,楊潔,
(天津醫科大學1.生物化學系;2.基礎醫學研究中心;3.細胞生物學系,天津 300070)
論著
利用改良的CRISPR/Cas9基因編輯系統構建HeLa細胞SND1基因敲除穩定株
劉郁瑩1,崔曉騰1,高星杰2,趙秀娟3,付雪1,葛林1,蘇超2,楊潔1,2
(天津醫科大學1.生物化學系;2.基礎醫學研究中心;3.細胞生物學系,天津 300070)
目的:利用改良的CRISPR/Cas9基因編輯系統敲除HeLa細胞中SND1基因,構建HeLa細胞SND1基因敲除穩定株。方法:設計一對特異性識別SND1基因第二個啟動子的上下游sgRNA,以PX462質粒為載體,構建出一對重組真核表達質粒。酶切和測序鑒定后,將一對重組質粒共同轉染進入HeLa細胞中,使用嘌呤霉素進行陽性細胞篩選,挑取單克隆細胞進行培養。最后用Western Blot鑒定敲除效果。結果:sgRNA正確插入到PX462質粒載體中,轉染并篩選單克隆后的細胞中沒有SND1蛋白的表達。結論:成功構建出HeLa細胞SND1基因敲除穩定株。
SND1;基因敲除;CRISPR/Cas9;HeLa細胞
人類SND1(Staphylococcal Nuclease Domain-Containing Protein 1)蛋白是在不同種屬間具有高度保守性的蛋白質[1-4],廣泛參與細胞多種生物學過程[5-8]。本實驗是利用改良的CRISPR/Cas9基因編輯系統,即以一對單鏈向導RNA(single guide-RNA, sgRNA)靶標SND1基因,招募Cas9核酸酶的突變體Cas9n核酸切口酶(Cas9 D10 nickase,Cas9n)對其進行切割,從而敲除SND1基因,構建出HeLa細胞SND1基因敲除穩定株,為更加深入地研究SND1蛋白的功能提供了一個SND1基因敲除的人類細胞模型。
1 材料與方法
1.1 試劑及儀器 HeLa細胞為本實驗室留存;真核表達質粒pSpCas9n(BB)-2A-Puro(PX462)由天津醫科大學吳旭東教授惠贈;trans1-T1感受態細胞、Trans2K PlusII DNA marker購自北京全式金生物技術有限公司;限制性內切酶BbsI、DMEM高糖培養基購自Thermo公司;T4連接酶購自NEB公司;質粒保護的核酸外切酶(plasmid safe exonuclease)購自Epicentre公司;去內毒素質粒提取試劑盒、膠回收試劑盒購自Biomiga公司;Neofect轉染試劑購自零客創智生物科技有限公司;BCA蛋白測定試劑盒購自Pierce公司;鼠源SND1蛋白抗體為本實驗室自制[9];β-tubulin蛋白抗體購自Sigma公司;辣根過氧化物酶標記的抗鼠源IgG二抗購自Fermentas公司;LumiGLo化學發光底物購于KPL公司;RIPA裂解液、嘌呤霉素(puromycin)購自北京索萊寶科技有限公司。CO2培養箱購自Thermo公司;Western blot電泳槽、電泳儀購自Bio-Rad公司。sgRNA序列合成由蘇州金唯智生物科技有限公司完成;質粒測序由北京天一輝遠生物科技有限公司完成。
1.2 實驗方法
1.2.1 針對SND1基因序列的sgRNA設計 (1)采用http://www.ncbi.nlm.nih.gov/網站確定人類SND1(NM_014390.2)的基因序列。(2)根據文獻提示[10],使用 http://crispr.mit.edu/設計 SND1基因 double nick的上下游sgRNA。sgRNA設計原則:(1)sgRNA的5′端第一個堿基若不是G,則需要在前面加上1個G;(2)以sgRNA序列為模版,設計出其互補鏈;(3)sgRNA及其互補鏈分別添加BbsI的酶切位點。
1.2.2 重組真核表達質粒PX462-sgRNA的構建 (1)PX462載體線性化:使用快速限制性內切酶BbsI線性化PX462質粒載體,切膠回收。體系:1 μg PX462載體,1 μL BbsI,1 μL FastAP,2 μL 10×Green FastDigest Buffer,加水稀釋至20 μL。程序:37℃,30 min;4℃,stop。(2)sgRNA的合成及形成二聚體:人工合成sgRNA寡核苷酸鏈A、B及其互補鏈,使用降落PCR退火形成二聚體。體系:100 nmol sgRNA-A/B,100 nmol sgRNA-A/B的互補鏈,1 μL 10×T4 Ligation Buffer,0.5 μL T4 PNK,加水至10 μL。程序:37℃,30 min;95℃,5 min;-1℃/min,至25℃;4℃,stop。(3)sgRNA二聚體與PX462線性載體連接:PX462載體和降落PCR產物比例為1∶3,室溫(25℃)反應60min。體系:PX462線性載體50ng,降落PCR產物150 ng,1 μL 10×T4 Ligation Buffer,加水稀釋至10 μL,1 μL T4 Ligase。程序:25℃,60 min;4℃,stop。(4)重組質粒純化:使用質粒保護的核酸外切酶去除非特異連接。體系:11μL二聚體與載體連接后的產物,1.5 μL 10×PlasmidSafe Buffer,1.5 μL 10mmolATP,1μLPlasmidSafeexonuclease。程序:37℃,30 min;4℃,stop。(5)轉化trans1-T1感受態細胞,挑取單克隆搖菌,使用去內毒素質粒小提試劑盒提取質粒,進行酶切和測序驗證。
1.2.3 重組真核表達質粒的酶切鑒定 使用快速限制性內切酶BbsI對PX462質粒載體、PX462-sgRNA-A、PX462-sgRNA-B進行酶切。體系:1 μg質 粒 ,1 μL BbsI,1μL FastAP,2 μL 10×Green FastDigest Buffer,加水稀釋至20 μL。程序:37℃,30 min;4℃,stop。酶切后,使用1%的瓊脂糖凝膠電泳對酶切產物進行鑒定。
1.2.4 HeLa細胞培養及轉染 以去內毒素質粒提取試劑盒提取無內毒素質粒載體PX462和重組真核表達質粒PX462-sgRNA-A、PX462-sgRNA-B。 HeLa細胞接種至6 cm皿中,加入含10%胎牛血清的DMEM高糖培養基,置于37℃、5%的CO2培養箱中培養,至細胞50%~60%匯合時,根據產品說明書使用Neofect轉染試劑瞬時轉染質粒載體PX462(HeLa-SND1-WT)或共轉重組真核表達質粒PX462-sgRNA-A和 PX462-sgRNA-B(HeLa-SND1-KO)。
1.2.5 HeLa細胞SND1基因敲除穩定株的單克隆篩選 轉染后48 h,更換培基為含有2 μg/mL Purom ycin和10%胎牛血清的DMEM高糖培養基,以未轉染的HeLa細胞為陰性對照。加藥48 h后,未轉染的HeLa細胞全部被殺死。72 h后撤去培基中的Puromycin繼續培養。待細胞長滿,將細胞用胰酶消化,鋪到10 cm皿中,平均每個顯微鏡低倍鏡視野中有3~5個細胞即可。待細胞長成單克隆集落,用槍頭分別將單克隆集落輕輕刮取,移至96孔板中培養。待96孔板長滿,移至24孔板中繼續培養。1.2.6 HeLa細胞SND1基因敲除穩定株Western Blot檢測 將篩選后的單克隆穩定株HeLa-SND1-WT和HeLa-SND1-KO分別傳至6 cm皿中培養。待細胞長滿,使用預冷的PBS緩沖液對細胞洗滌3次,吸凈PBS后,加入300μL RIPA裂解液,用干凈的細胞刮刮取細胞至1.5 mL離心管中,冰上靜置裂解15 min。之后對其進行超聲處理,強度60%,時間30 s(1次10 s,間隔5 s,共進行3次)。4℃13 000 r/min超速離心10 min,取上清細胞裂解液。用BCA蛋白測定試劑盒測定裂解液的蛋白濃度后,加入5×上樣緩沖液(50%甘油,10%SDS,0.5%溴酚藍,250 mmol pH6.8 Tris-HCl),99℃對其熱變性10 min。進行8% SDS-PAGE電泳后,用濕電轉膜儀將PAGE膠上蛋白質轉移到0.45 μm PVDF上。使用含有5%脫脂牛奶的TBST溶液封閉2 h后,用自制鼠源SND1抗體4℃孵育過夜。使用TBST溶液洗滌PVDF膜3次,每次10 min。用辣根過氧化物酶標記的抗鼠源IgG二抗室溫孵育2 h。之后再用TBST溶液洗滌PVDF膜3次,每次10 min。孵育LumiGLo化學發光底物1 min,進入暗室曝光。
2 結果
2.1 sgRNA靶點的選擇及寡核苷酸鏈的設計 由于SND1基因第1個外顯子區(expressed region 1, EXON 1)和基因編碼區(Coding Sequence,CDS)的重疊區域過短,因此選取第2個外顯子區進行sgRNA設計。CRISPR/Cas9系統工作示意圖如圖1。設計好的sgRNA及其互補鏈如表1。
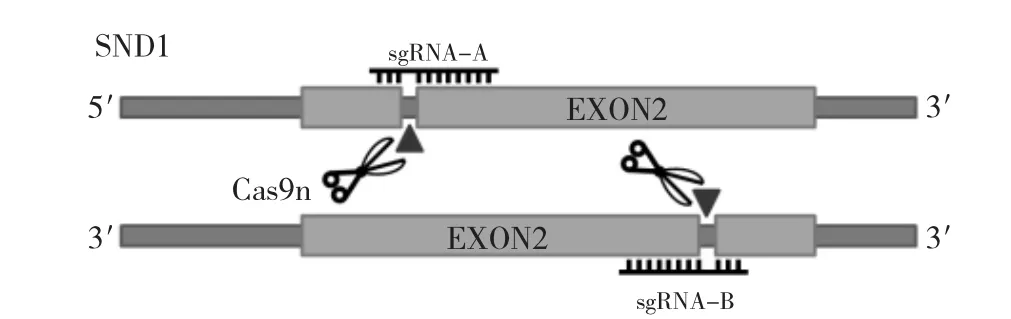
圖1 CRISPR/Cas9系統切割SND1基因外顯子上下游Fig 1 CRISPR/Cas9 system cut the upstream and downstream of SND1

表1 上下游sgRNA及其互補鏈寡核苷酸序列Tab 1 The nucleotide sequences of the upstream and downstreamsgRNA
2.2 重組真核表達質粒PX462-sgRNA的酶切和測序結果 利用BbsI限制性內切酶對構建好的PX462-sgRNA-A和PX462-sgRNA-B進行酶切。結果如圖2 A,成功構建的質粒不含有BbsI酶切位點,因此不能被切割為線性。測序結果如圖2 B,插入序列的位置、方向均正確,質粒構建成功。
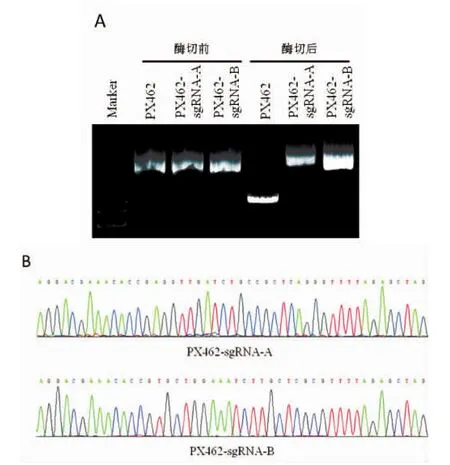
圖2 重組真核表達質粒的酶切和測序結果Fig 2 The enzyme digestion and sequencing of recombinant eukaryotic expressional plasmid
2.3 Western blot檢測HeLa細胞SND1基因敲除穩定株中SND1蛋白的表達 結果如圖3,同HeLa-SND1-WT相比,HeLa-SND1-KO中的SND1沒有表達,HeLa細胞SND1基因敲除穩定株構建成功。
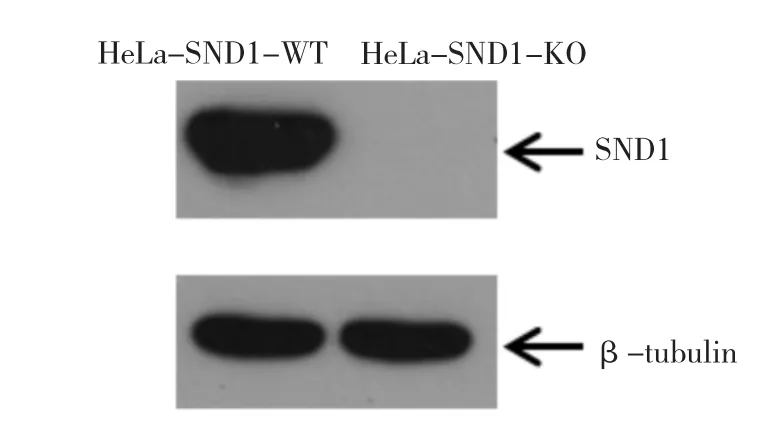
圖3 單克隆穩定株Western blotFig 3 Western blotting of monoclonal stable strain
3 討論
SND1蛋白序列在不同生物中具有高度的保守性,參與調控細胞多種重要的生理過程。有研究表明,SND1蛋白在腫瘤遷移中也有作用[11]。因此,建立穩定敲除SND1蛋白的細胞株有助于研究其生理病理條件下的功能。
本實驗中,我們利用CRISPR/Cas9系統來完成對HeLa細胞中SND1基因的靶向性敲除。CRISPR(clustered regularly interspaced short palindromic repeat sequences,CRISPR)序列本是細菌抵御病毒入侵的防御機制,近年來科學家對其進行改造,成為基因編輯的又一有力武器。傳統CRISPR/Cas9系統是以sgRNA為向導,靶標目的基因,招募Cas9核酸酶對DNA雙鏈進行切割,從而敲除該基因[12]。對比 ZFN(Zinc-finger nucleases) 技術和 TALEN(transcription activator-like effector nucleases)技術,此方法簡便易行,成為靶向基因編輯的重要技術。但是由于sgRNA長度的局限,傳統CRISPR/Cas9系統容易產生脫靶效應,即sgRNA識別到其他基因處,導致其他基因敲除。麻省理工學院的Zhang等[13]對其進行改良,將Cas9核酸酶的RuvC結構域中的天冬氨酸突變成丙氨酸(D10A),導致RuvC結構域失去活性,突變后的Cas9酶變成Cas9n核酸切口酶,只能切割單鏈。如果DNA只被一個Cas9n切割,產生單鏈缺口,細胞很快利用堿基切除修復途徑進行DNA修復。因此使用一對sgRNA對目的基因上下游進行靶標并招募Cas9n進行切割,導致細胞利用非同源性末端接合(non-homologous end joining,NHEJ)機制進行DNA修復。此修復機制并不精確,易產生框移突變,從而達到敲除目的基因的作用。
實驗中,我們選擇PX462質粒作為載體,該質粒可以同時表達Cas9n和插入的sgRNA,簡化了表達Cas9n質粒和sgRNA質粒共轉染的步驟。轉染后,使用2 μg/mL Puromycin進行陽性細胞篩選。同時使用未轉染的HeLa細胞作為對照,也加入2 μg/mL Puromycin,待該細胞全部殺死時,即認為轉染組存活下來的細胞為帶有Puromycin抗性的陽性細胞。之后撤掉藥物,使用完全培基對其進行培養和單克隆篩選。最后,使用WesternBlot對單克隆細胞進行鑒定,從而獲得HeLa細胞SND1基因敲除穩定株。
[1] Saarikettu J,Ovod V,Vuoksio M,et al.Monoclonal antibodies against human Tudor-SN[J].Hybridoma(Larchmt),2010,29(3):231
[2] Rodríguez L,Ochoa B,Martínez M J.NF-Y and Sp1 are involved in transcriptional regulation of rat SND p102 gene[J].Biochem Biophys Res Commun,2007,356(1):226
[3]Broadhurst M K,Lee R S,Hawkins S,et al.The p100 EBNA-2 coactivator:a highly conserved protein found in a range of exocrine and endocrine cells and tissues in cattle[J].Biochim Biophys Acta, 2005,1681(2/3):126
[4] Zhao C T,Shi K H,Su Y,et al.Two variants of zebrafish p100 are expressed during embryogenesis and regulated by Nodal signaling [J].FEBS Lett,2003,543(1/3):190
[5] Liu X,Dong L,Zhang X,et al.Identification of p100 target promoters by chromatin immunoprecipitation-guided ligation and selection(ChIP-GLAS)[J].Cell Mol Immunol,2011,8(1):88
[6] Gao X,Zhao X,Zhu Y,et al.Tudor staphylococcal nuclease(Tudor-SN)participates in small ribonucleoprotein (snRNP)assembly via interacting with symmetrically dimethylated Sm proteins[J].J Biol Chem,2012,287(22):18130
[7] Su C,Zhang C,Tecle A,et al.Tudor staphylococcal nuclease (Tudor-SN),a novel regulator facilitating G1/S phase transition, acting as a co-activator of E2F-1 in cell cycle regulation[J].J Biol Chem,2015,290(11):7208
[8] Gao X,Fu X,Song J,et al.Poly(a)(+)mRNA-binding protein Tudor-SN regulates stress granules aggregation dynamics[J].FEBS J,2015,282(5):874
[9] Saarikettu J,Ovod V,Vuoksio M,et al.Monoclonal antibodies against human Tudor-SN[J].Hybridoma(Larchmt),2010,29(3):231
[10]Ran F A,Hsu P D,Wright J,et al.Genome engineering using the CRISPR-Cas9 system[J].Nat Protoc,2013,8(11):2281
[11]Yu L,Liu X,Cui K,et al.SND1 Acts downstream of TGFβ1 and upstream of smurf1 to promote breast Cancer metastasis[J].Cancer Res,2015,75(7):1275
[12]Gilbert L A,Larson M H,Morsut L,et al.CRISPR-Mediated modular RNA-Guided regulation of transcription in eukaryotes[J]. Cell,2013,154(2):442
[13]Ran F A,Hsu P D,Lin C Y,et al.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity[J].Cell,2013, 154(6):1380
(2015-04-13收稿)
Construction of HeLa SND1 knockout gene stable strain by using modified CRISPR/Cas9 gene editing system
LIU Yu-ying1,CUI Xiao-teng1,GAO Xing-jie2,ZHAO Xiu-juan3,FU Xue1,GE Lin1,SU Chao2,YANG Jie1.2
(Tianjin Medical University 1.Department of Biochemistry;2.Research Center of Basic Medical Science;3.Department of Cell Biology, Tianjin 300070,China)
Objective:To apply modified CRISPR/Cas9 gene editing system to knock out the SND1 gene in HeLa cell and construct HeLa SND1 gene knockout stable strain.Methods:A pair of sgRNAs that could specially identify the upstream and downstream of SND1 gene second promoter were designed,then a recombinant eukaryotic expressional plasmid by the carrier of PX462 was constructed.After enzyme digestion and sequencing,a pair of recombinant plasmids into HeLa cell were co-transfected,then puromycin was used to screen positive cell and the monoclonal cell was developed.The knockout effect was measured by western blotting.Results:sgRNA was correctly inserted into the PX462 recombinant plasmid,and SND1 protein was undetected in HeLa cell after transfection and screening of monoclonal cell.Conclusion:HeLa SND1 gene knockout stable strain can be successfully built.
SND1;gene knockout;CRISPR/Cas9;HeLa cells
Q7
A
1006-8147(2015)06-0480-04
劉郁瑩(1986-),女,碩士在讀,研究方向:腫瘤免疫;通信作者:楊潔,E-mail:yangji@tmu.edu.cn。
