銅綠假單胞菌clpP基因缺陷株的構建
2015-03-17 01:46:58張加勤饒慧華徐巧麗黃朝陽房麗麗馬曉波宋秀宇
中國人獸共患病學報 2015年7期
張加勤,饒慧華,2,徐巧麗,2,黃朝陽,房麗麗,馬曉波,宋秀宇
銅綠假單胞菌clpP基因缺陷株的構建
張加勤1,饒慧華1,2,徐巧麗1,2,黃朝陽1,房麗麗1,馬曉波1,宋秀宇3
目的 構建銅綠假單胞菌clpP基因缺陷株。方法 分別以質粒pUCGM和銅綠假單胞菌PAO1基因組為模板PCR擴增慶大霉素抗性基因(GM)和銅綠假單胞菌clpP基因及其3′、5′側翼序列,將clpP基因及其3′、5′側翼序列克隆至pMD19T載體,EcoRV切除clpP基因37 bp~453 bp片段后,引入慶大霉素抗性基因,構建重組質粒pCKR2;EcoRⅠ/HindⅢ雙酶切重組質粒pCKR2,回收FclpP-GM-clpPR片段,與自殺質粒pEX18Tc連接,得到clpP基因缺陷的同源重組載體pCKR3;將pCKR3質粒轉化大腸埃希菌SM10,與銅綠假單胞菌PAO1雙親雜交,慶大霉素篩選得到銅綠假單胞菌clpP基因缺陷株。結果 經酶切鑒定同源重組載體pCKR3構建正確;PCR和DNA測序鑒定銅綠假單胞菌clpP基因缺陷株構建成功。結論 本研究成功敲除了銅綠假單胞菌clpP基因,為進一步研究clpP基因的生物學功能奠定了基礎。
銅綠假單胞菌;clpP基因;雙親雜交;同源重組
銅綠假單胞菌(Pseudomonasaeruginosa,P.aeruginosa)是一種革蘭陰性條件致病菌,能夠在土壤、海洋以及動植物的組織等多種環境中生長繁殖,可引起菌血癥、泌尿道感染和醫院獲得性肺炎。其中,銅綠假單胞菌所致的肺部慢性感染是肺部囊性纖維化病人高發病率和高死亡率的主要原因[1]。細菌定植及致病過程中,易受到來自機體免疫系統的攻擊,引起菌體內蛋白質的損傷及異常聚集,如不及時清除會對細菌造成致命的損傷。研究表明,在熒光假單胞菌、大腸埃希菌及低GC含量革蘭氏陽性細菌中,ClpP蛋白酶在細菌清除不可逆損傷蛋白質、維持內環境穩態等生物過程中發揮著重要作用[2-6]。銅綠假單胞菌ClpP蛋白酶由clpP基因編碼,基因大小642 bp,與熒光假單胞菌clpP基因同源性高達84%,編碼產物ClpP蛋白酶與熒光假單胞菌同源性達81%。因此我們推斷銅綠假單胞菌ClpP蛋白酶亦具有與熒光假單胞菌ClpP蛋白酶相似的生物學功能。然而,目前關于銅綠假單胞菌ClpP蛋白酶功能及結構的報道較少,為研究ClpP在銅綠假單胞菌中的生物學功能,我們采用同源重組的方法構建了銅綠假單胞菌clpP基因缺陷株。
1 材料與方法
1.1 菌株和質粒 銅綠假單胞菌PAO1、大腸埃希菌DH5α、大腸埃希菌SM10為本實驗室保存。質粒pMD19T 購自TaKaRa公司;pUCGM和pEX18Tc 載體為本實驗室保存。
1.2 主要試劑和儀器 DNA 連接酶、限制性內切酶購自TaKaRa公司;PfuDNA Polymerase、質粒提取試劑盒購自上海生工生物工程技術服務有限公司;2×Taq PCR MasterMix、細菌基因組提取試劑盒購自北京天根生化科技公司;PCR產物/凝膠回收純化試劑盒購自QIAGEN公司;其余為國產或進口分析純試劑;PCR引物合成及DNA測序均由上海立菲生物技術有限公司完成。
1.3 實驗步驟
1.3.1 同源重組載體pCKR3的構建
1.3.1.1clpP基因及其3′、5′側翼序列和GM片段的克隆及鑒定 采用細菌基因組提取試劑盒提取銅綠假單胞菌PAO1基因組,設計PCR引物:clpP-F1(5′-CTGCCGAGTTCACCGTTAC-3′)和clpP-R1(5′-TCCTTCTTGTCACGCTGGT-3′),以P. aeruginosa PAO1基因組為模板擴增clpP基因及其3′、5′側翼序列。反應體系為25 μL,反應條件為:95 ℃預變性5 min; 95 ℃ 30 s,57 ℃ 30 s,72 ℃ 1 min,共30個循環;72 ℃延伸10 min。采用質粒提取試劑盒提取pUCGM質粒DNA ,設計PCR引物:GM-F1(5′-CTCGAATTGACATAAGCCTG-3′)和GM-R1(5′-TACATTATACGAACGGTACG-3′),以pUCGM為模板,采用PfuDNA Polymerase擴增慶大霉素抗性基因。反應體系為50 μL,反應條件為:95 ℃預變性5 min;95 ℃ 30 s,57 ℃ 30 s,72 ℃ 2 min,共30個循環;72 ℃延伸10 min。擴增產物均經1%瓊脂糖凝膠電泳鑒定,PCR回收純化試劑盒回收,至-20 ℃保存備用。
1.3.1.2 重組質粒pCKR1的構建clpP基因及其3′、5′側翼序列克隆至pMD19T載體上,轉化大腸埃希菌DH5α感受態細胞,氨芐青霉素(100 μg/mL)篩選陽性克隆,37 ℃過夜培養,提取質粒用EcoRV酶切鑒定,重組質粒命名為pCKR1。
1.3.1.3 重組質粒pCKR2的構建 限制性內切酶EcoRV酶切pCKR1,電泳,回收大片段,與GM片段連接,慶大霉素(10 μg/mL)篩選獲得重組載體pCKR2。
1.3.1.4 同源重組載體pCKR3的構建 限制性內切酶EcoRⅠ/HindⅢ雙酶切pCKR2,回收FclpP-GM-clpPR片段和EcoRⅠ/HindⅢ酶切的pEX18Tc連接,慶大霉素(10 μg/mL)和四環素(100 μg/mL)篩選獲得重組載體pCKR3。將pCKR3轉化大腸埃希菌SM10感受態,命名為SCKR1,提取質粒用EcoRⅠ和HindⅢ酶切鑒定。
1.3.2 雙親雜交構建clpP缺陷株 銅綠假單胞菌PAO1與大腸埃希菌SCKR1培養至OD600分別為0.8~1.0和0.6~0.8,收集并洗滌菌體,以1∶4的比例混合于200 μL LB中,滴加于固相濾膜上,繼續培養24 h。刮取固相膜上的菌落,洗滌并重懸,涂布于含有慶大霉素(40 μg/mL)的抗性平板上,37 ℃培養48 h。挑取陽性克隆經含慶大霉素的營養肉湯增菌培養,提取基因組DNA,PCR分析和DNA測序鑒定,引物為clpP-F2(5′-CCTGGTGGTTGCTCAGTTGCT-3′)/clpP-R2(5′-GCCGATACAGGTGGTCGAGACG-3′)。
2 結 果
2.1clpP基因和GM抗性片段的克隆與鑒定 以clpP-F1和clpP-R1為引物擴增clpP基因及其3′、5′側翼序列,產物大小1.7 kb;以GM-F1和GM-R1為引物擴增的GM抗性片段大小約0.9 kb(圖1)。
2.2 重組質粒pCKR1的鑒定clpP基因及其3′、5′側翼序列與載體pMD19T連接,轉化大腸埃希菌DH5α,EcoRV酶切鑒定,得到小片段0.4 kb和大片段4.0 kb(圖2),測序結果與目的片段序列一致。
2.3 重組質粒pCKR2的鑒定 GM片段與pCKR1的EcoRV酶切大片段連接,構建重組質粒pCKR2。pCKR2經EcoRⅠ和HindⅢ酶切后分別得到2.3 kb的短片段和2.6 kb的長片段(圖3),測序結果與目的片段序列一致。
2.4 同源重組載體pCKR3的鑒定 回收pCKR2經EcoRⅠ/HindⅢ酶切的FclpP-GM-clpPR片段,建立與EcoRⅠ/HindⅢ酶切的自殺質粒pEX18Tc的連接體系,獲得同源重組載體pCKR3。pCKR3經EcoRⅠ和HindⅢ酶切鑒定,結果出現兩條大小各為2.3 kb和6.2 kb的條帶(圖4),測序結果與目的片段序列一致。

M: D2000 marker; 1: PCR product of GMRcassette; 2: PCR product ofclpPgene with 3′ and 5′ flanking sequences.
圖1 慶大霉素抗性基因和clpP基因及其3′、5′側翼序列的PCR結果
Fig.1 PCR products of gentamicin resistant cassette andclpPgene with 3′ and 5′ flanking sequences
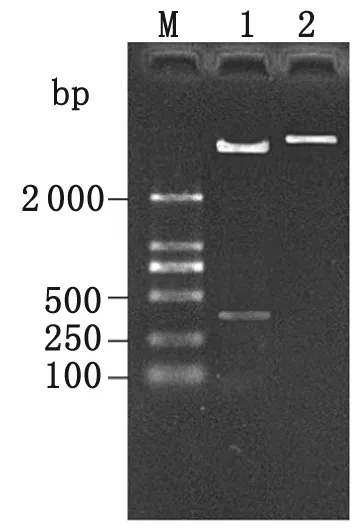
M: D2 000 marker; 1:EcoRV digestion of recombinant plasmid pCKR1; 2: Recombinant plasmid pCKR1
圖2 重組質粒pCKR1的酶切鑒定結果
Fig.2 Restriction enzyme digestion of the recombinant plasmid pCKR1
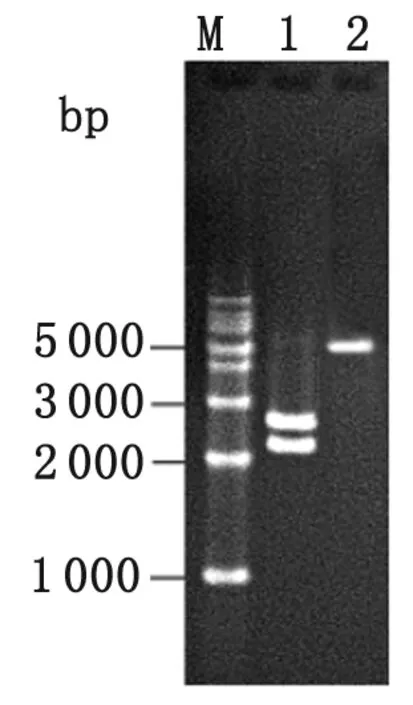
M: 1 kb marker; 1:EcoRⅠ/HindⅢ digestion of recombinant plasmid pCKR2; 2: recombinant plasmid pCKR2.
圖3 重組質粒pCKR2的酶切鑒定結果
Fig.3 Restriction enzyme digestion of the recombinant plasmid pCKR2
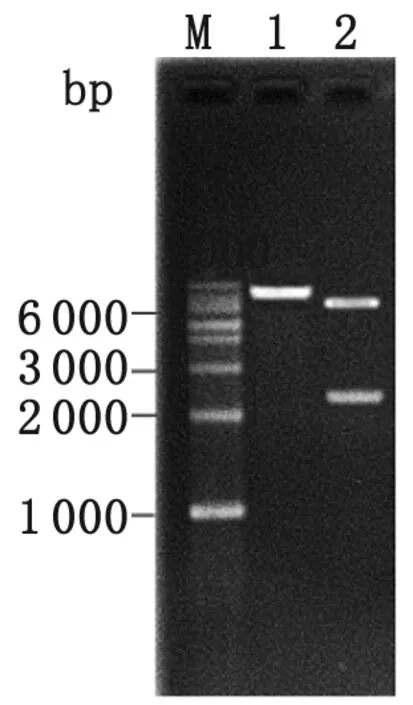
M: 1 kb marker; 1: Homologous recombinant plasmid pCKR3; 2:EcoRⅠ/HindⅢ digestion of pCKR3
圖4 同源重組載體pCKR3的酶切鑒定結果
Fig.4 Restriction enzyme digestion of the homologous recombinant vector pCKR3
2.5 含有慶大霉素抗性的銅綠假單胞菌clpP缺陷株的鑒定 大腸埃希菌SCKR1與銅綠假單胞菌PAO1雙親雜交后,陽性轉化株可在含有慶大霉素的培養基中生長良好,說明慶大霉素基因成功整合至銅綠假單胞菌的基因組中,并正確表達。PCR鑒定結果顯示,以缺陷株基因組為模板不能擴增出clpP基因片段(圖5),DNA測序顯示clpP基因部分序列已被刪除,且含有慶大霉素抗性基因。
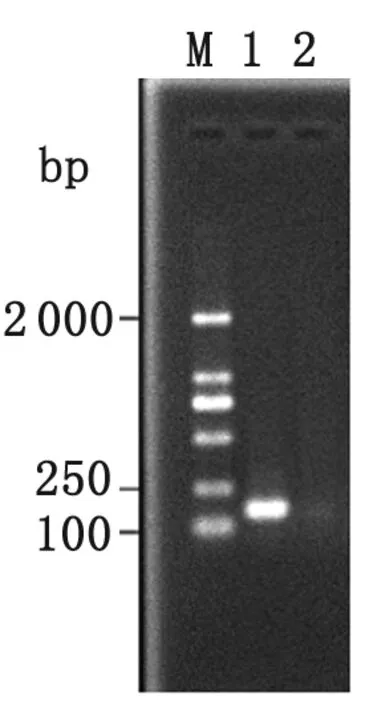
M: D2000 marker; 1: PCR product ofP.aeruginosaPAO1; 2: PCR product ofclpPmutant
圖5 銅綠假單胞菌clpP基因突變株的PCR鑒定結果
Fig.5 Electrophoresis ofP.areuginosamutation with deletedclpPgene
3 討 論
構建基因功能缺失株和過表達株是后基因組時代研究基因功能最直接和最有效的方法之一。同源重組技術自20世紀70年代建立之初,即成為修飾真核、原核基因組遺傳信息的常規手段,該技術可使外源基因整合入靶基因組上某一特定位點,構建目的基因缺失突變株。為研究銅綠假單胞菌ClpP蛋白酶的生物學功能,本研究采用同源重組染色體基因修飾技術構建銅綠假單胞菌clpP基因缺陷株,具有準確、高效的特點。同源重組的準確性與同源重組載體的同源度及同源臂大小密切相關,本研究在目的基因的上下游各設計500 bp ~ 1 000 bp的同源臂,既能滿足重組的高度準確性,又不至于使重組片段過大而影響其進入菌體的數量。
為提高重組效率,本研究采用pEX18Tc質粒作為同源重組片段的載體,該質粒是適用于銅綠假單胞菌的自殺質粒,帶有四環素抗性基因,由于銅綠假單胞菌中不含有pEX18Tc復制起始所需的復制蛋白,自殺質粒在細胞內不能復制,在外界選擇性壓力作用下,自殺質粒載體所攜帶的突變了的目的基因與宿主染色體基因組發生同源重組,利用目的基因內部插入的抗性基因篩選得到目的基因缺失突變株,而質粒載體本身由于自殺特性會隨著細菌的傳代從菌體內消失。采用自殺質粒不僅方便篩選,而且易于獲得高濃度的重組片段,提高重組效率。
以往通常將質粒用化學或電轉化的方法導入受體菌,兩種方法轉化效率雖高,但對于銅綠假單胞菌的轉化條件要求十分嚴格。因為銅綠假單胞菌在其培養過程中會產生許多胞外多糖基質、纖維蛋白和脂質蛋白,包裹著菌體外層[7],采用化學方法細胞難以形成感受態,影響轉化效率;若采用電轉化,需選擇較大的電壓和較長的時間,但電壓過大往往導致細胞死亡過度,電壓不足時,在靶細胞壁上形成的親水通道又不足以維持外源DNA分子的進入,導致轉化效率低下。因此我們采用接合轉移,雙親雜交是以含有F因子的革蘭陰性菌作為供體菌,將自身含有的質粒轉移至受體菌中的一種接合轉移。大腸埃希菌SM10包含pir基因,可用于質粒克隆和接合。以SM10為供體菌,銅綠假單胞菌為受體菌,可將構建好的質粒例如pCKR3轉移至銅綠假單胞菌,完成同源重組工作。接合轉移不需要制作感受態細胞,操作簡便,適用于銅綠假單胞菌的轉化。本研究采用接合轉移已成功構建了銅綠假單胞菌clpP基因缺陷株,并通過測序鑒定證明構建正確,這將為下一步研究clpP基因的具體生物學功能奠定基礎。
[1]Qiu D, Eisinger VM, Head NE, et al. ClpXP proteases positively regulate alginate overexpression and mucoid conversion inPseudomonasaeruginosa[J]. Microbiology, 2008,154(Pt 7): 2119-2130.
[2]Dougan DA, Mogk A, Bukau B. Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question[J]. Cell Mol Life Sci, 2002, 59(10): 1607-1616.
[3]Gottesman S. Proteases and their targets inEscherichiacoli[J]. Annu Rev Genet, 1996, 30: 465-506.
[4]WANG zhen-hai,SUN ye-qing. Progress in Study on Clp Protease[J]. Pharmaceutical Biotechnology,2005,12(6):412-415. 王振海,孫野青. Clp蛋白酶研究進展[J]. 藥物生物技術, 2005,12(6):412-415.英文對照
[5]Yu AY, Houry WA. ClpP: a distinctive family of cylindrical energy-dependent serine proteases[J]. FEBS Lett, 2007, 581(19): 3749-3757.
[6]de Bruijn I, Raaijmakers JM. Regulation of cyclic lipopeptide biosynthesis inPseudomonasfluorescensby the ClpP protease[J]. J Bacteriol, 2009, 191(6): 1910-1923.
[7]Bjarnsholt T, Jensen PO, Fiandaca MJ, et al.Pseudomonasaeruginosabiofilms in the respiratory tract of cystic fibrosis patients[J]. Pediatr Pulmonol, 2009, 44(6): 547-558.
Construction of theclpPmutant strain ofPseudomonasaeruginosa
ZHANG Jia-qin1,RAO Hui-hua1,2,XU Qiao-li1,2,HUANG Chao-yang1,FANG Li-li1,MA Xiao-bo1,SONG Xiu-yu3
(1.DepartmentofClinicalLaboratory,theFirstAffiliatedHospitalofXiamenUniversity,Xiamen361003,China;2.TheFirstClinicalMedicalCollegeofFujianMedicalUniversity,Fuzhou350005,China;3.XiamenCentralBloodServiceStation,Xiamen361003,China)
To deleteclpPgene ofPseudomonasaeruginosaPAO1, theclpPgene with 3′ and 5′ flanking sequences was amplified by PCR using the primers clpP-F1and clpP-R1, and then the PCR product was cloned into pMD19T for construction of pCKR1. A gentamicin resistance cassette amplified from pUCGM was inserted intoEcoRV sites of pCKR1 for construction of pCKR2. Then the FclpP-GM-clpPR fragment was obtained by cutting pCKR2 withEcoRⅠ/HindⅢ and gel purification, and inserted it intoEcoRⅠ/HindⅢ sites of pEX18Tc to construct the homologous recombinant vector pCKR3. TheP.aeruginosaclpPmutant strain was obtained by conjugations betweenP.aeruginosaPAO1 andE.coliSM10 transformed with pCKR3. The homologous recombinant vector pCKR3 was verified by enzymatic digestion and the finalP.aeruginosaclpPmutant strain was verified by PCR analysis and DNA sequencing. TheclpPgene ofP.aeruginosawas successfully deleted, which laid a foundation for further study of its biological function.
Pseudomonasaeruginosa;clpPgene; conjugations; homologous recombinant
Song Xiu-yu, Email: songxyxm@126.com
國家自然科學基因(No.81000762);福建省自然科學基金(No.2010D018,2013D002)
宋秀宇,Email:songxyxm@126.com
1.廈門大學附屬第一醫院檢驗科,廈門 361003; 2.福建醫科大學第一臨床醫學院,福州 350005; 3.廈門市中心血站,廈門 361003
10.3969/cjz.j.issn.1002-2694.2015.07.007
R378
A
1002-2694(2015)07-0627-04
2014-09-09;
2015-05-01

Supported by the National Natural Science Fundation (No. 81000762) and the Natural Science Fund of Fujian Province (Nos. 2010D018 and 2013D002)
