火焰原子吸收光譜法測定錸產品中的痕量鈉
2015-04-18 03:17:01慧舒波周冬梅張選冬華宏全楊坤彬史誼峰代紅坤王傳飛鄭文英李君房勇吳志清林波劉文朱利亞
中國無機分析化學 2015年4期
關鍵詞:實驗
唐 慧舒 波周冬梅張選冬華宏全楊坤彬史誼峰代紅坤王傳飛鄭文英李 君房 勇吳志清林 波劉 文朱利亞*
(1云南銅業股份有限公司,昆明650102;2貴研資源(易門)有限公司稀貴金屬綜合利用新技術國家重點實驗室,昆明650106)
火焰原子吸收光譜法測定錸產品中的痕量鈉
唐 慧1舒 波1周冬梅1張選冬2華宏全1楊坤彬1史誼峰1代紅坤1王傳飛1鄭文英1李 君1房 勇1吳志清1林 波2劉 文2朱利亞2*
(1云南銅業股份有限公司,昆明650102;2貴研資源(易門)有限公司稀貴金屬綜合利用新技術國家重點實驗室,昆明650106)
建立了火焰原子吸收光譜法測定高錸酸、高錸酸銨、錸粉中痕量鈉的方法,對樣品的預處理和測定鈉的條件進行了研究。結果表明:水溶解法、硝酸溶解法或硝酸-硫酸溶解法溶解樣品完全,測得鈉的結果吻合;于選定條件下,鈉的測定濃度在0.020 0~0.500 0μg/mL范圍內線性良好;測定高錸酸、高錸酸銨和錸粉樣品中0.27~0.47mg/L、0.000047%~0.00048%和0.000040%~0.00049%的鈉含量,檢出限、相對標準偏差(RSD,n=7)、加標回收率分別為高錸酸3×10-4μg/mL、6%~10%、98%~102%,高錸酸銨3×10-4μg/mL、8%~9%、96%~102%和錸粉3×10-4μg/mL、5%~9%、96%~103%。方法結果準確、分析快速、操作簡便,應用于實際的樣品分析,結果滿意。
鈉;痕量;錸產品;火焰原子吸收光譜法
0 前言
稀散金屬錸及其化合物產品是生產合金、耐高溫涂層、催化劑等的前驅體,在國防、航天航空、核能、電子和石油化工等高新技術領域中廣泛應用[12]。目前,錸及其化合物的初級產品主要從二次資源廢料中提取和制備,在此過程中將引入大量的鈉離子,致使純產品中仍含有痕量的雜質元素鈉,而其是影響后期生產的合金、耐高溫涂層、催化劑等產品質量的重要因素之一,故準確測定雜質元素鈉含量對控制產品質量是非常必要的。
關于其它樣品中鈉含量測定的方法,除有少量的電感耦合等離子體原子發射光譜法[3]、石墨爐原子吸收光譜法[4]和離子色譜法[5]介紹外,火焰原子吸收法光譜法因對鈉離子具有選擇性好、靈敏度高、吸光度穩定和線性范圍寬等優點,而成為主要的分析技術之一,并已廣泛用于純物質[6]、化合物[7-8]、合金[9]、氧化物[10]等[11-13]中。關于錸化合物中鈉和其它雜質元素的測定,標準[14]采用電感耦合等離子體原子發射光譜法直接測定高錸酸中0.5~50mg/L鈉含量,而標準[15]采用硝酸-硫酸溶解高錸酸銨,驅除基體錸對電感耦合等離子體原子發射光譜法測定0.0001%~0.010%鈹、鎂、鋁、鉀、鈣、鈦、鉻、錳、鐵、鈷、銅、鋅和鉬雜質元素含量的影響。作者曾采用電感耦合等離子體原子發射光譜法直接測定高錸酸、高錸酸銨和錸粉中的痕量鈉,觀察到因錸的發射強度較大,導致測得鈉含量明顯偏高。迄今未見到測定高錸酸、高錸酸銨和錸粉中痕量鈉的文獻報道。
本文研究了水溶解、硝酸溶解和硝酸-硫酸溶解樣品,以及原子吸收光譜法測定高錸酸、高錸酸銨和錸粉中痕量鈉的條件,對比了直接測定與驅除基體錸對鈉分析結果的影響。
1 實驗部分
1.1 主要試劑、儀器及材料
原子吸收光譜儀(SSERIES AA Spectrometer,美國熱電公司);分析天平(METTLER TOLEDO,德國);管理樣(編號,鈉參考值)為:高錸酸(C 1503,≤0.3mg/L),高錸酸銨(2014C26,≤0.00005%),錸粉(C 12M0401,≤0.00005%)。
鈉標準儲備溶液(1 000μg/mL,水介質):稱取0.254 21g基準氯化鈉(預先于105℃烘1h)于50mL石英燒杯中,加入少量水溶解,轉入100mL容量瓶中,用水稀釋至刻度,混勻(或采用國家標準溶液配制,國家鋼鐵材料測試中心鋼鐵研究總院)。
鈉標準溶液(A,10μg/mL):移取1.00mL鈉標準儲備溶液于100mL容量瓶中,用稀釋至刻度,混勻。
鈉標準溶液(B,0.20μg/mL):移取2.00mL鈉標準溶液(A)于100mL容量瓶中,用水稀釋至刻度,混勻。
乙炔氣:純度不小于99.999%;石英燒杯;石英容量瓶;石英表面皿;硝酸。試劑為優級純,水為超純水。
1.2 實驗方法
1.2.1 樣品的預處理
移取5.00mL高錸酸(C 1503)于10mL容量瓶中,用水稀釋至刻度,混勻;稱取約0.5g(精確至0.000 1g)高錸酸銨(2014C26)于50mL燒杯中,加入約5mL水,低溫溶解,取下,趁熱轉入10mL容量瓶中,冷卻后用水稀釋至刻度,混勻;稱取約0.5g(精確至0.000 1g)錸粉(C 12M0401)于50mL燒杯中,加入少量水潤濕,加入0.5mL硝酸,蓋上石英表面皿,于室溫反應停止后,再于低溫溶解,并蒸發至近干,加入約0.5mL水,蒸發至近干,重復一次。取下,冷卻,轉入10mL容量瓶中,用水稀釋至刻度,混勻。隨同樣品做空白實驗。
1.2.2 標準工作曲線的制作和測定
分別移取0、10.00mL鈉標準溶液(B),0.50、1.00、2.00、3.00、4.00、5.00mL鈉標準溶液(A)于100mL容量瓶中,用水稀釋至刻度,混勻,得到0、0.02、0.05、0.10、0.20、0.30、0.40、0.50μg/mL鈉標準級差溶液。
于表1儀器工作條件下,分別測定標準極差溶液、試液和空白實驗溶液,計算機擬合工作曲線,并給出試液中鈉的質量濃度,再依據稀釋倍數、定容體積和稱取樣品量,計算樣品中鈉的質量分數。推薦的分析線為鈉(Na)589.00nm。平行測定3次鈉標準級差溶液,平均吸光度為0、0.004、0.0102、0.0260、0.0514、0.0772、0.1029、0.1287。

表1 儀器工作條件Table 1 Instrumental operating condition
2 結果與討論
2.1 樣品溶解方法的選擇
采用管理樣高錸酸(C 1503)、高錸酸銨(2014C 26)、錸粉(C 12M0401)分別進行水溶解法、硝酸溶解法和硝酸-硫酸溶解法實驗。
2.1.1 水溶解法
高錸酸性狀為液體,可直接用水稀釋。稱取0.5g(精確至0.000 1g)高錸酸銨于50mL燒杯中,進行水溶解實驗。由實驗可知,至少10mL冷水可溶解高錸酸銨,但用該體積的冷水卻不易將其完全轉入容量瓶中,可采用小體積水加熱溶解后,趁熱完全轉入容量瓶中,冷卻后再用水稀釋至刻度。
2.1.2 硝酸溶解法
分別稱取約0.5g(精確至0.000 1g)高錸酸銨、錸粉于50mL燒杯中,加入0.5mL硝酸進行實驗。由實驗可知,高錸酸銨、錸粉均可快速、完全溶解于硝酸中,采用少量水蒸發至近干可驅除剩余氮氧化物。
2.1.3 硝酸-硫酸溶解法
移取5.00mL高錸酸于50mL燒杯中;分別稱取約0.5g(精確至0.000 1g)高錸酸銨、錸粉于50mL燒杯中,加入少量水潤濕。加入0.5mL硝酸、0.5 mL硫酸,蓋上表面皿,加熱至SO3白煙冒盡。取下,冷卻,加入0.5mL硫酸,1滴硝酸,蓋上表面皿,加熱至SO3白煙冒盡。取下,冷卻,加入少量水溶解鹽類,轉入10mL容量瓶中,用水稀釋至刻度,混勻。隨同樣品做空白實驗。由實驗可知,當硝酸-硫酸溶解或硫酸第一次發煙至小體積時,觀察到試液呈錸(Ⅶ)的特征淡黃色,當硫酸第二次發煙至小體積時,觀察到試液呈無色。表明錸(Ⅶ)已揮發除盡。
由表2可知,(1)水溶解法平均測得鈉含量、相對誤差分別為高錸酸0.27mg/L、0%~+4%,高錸酸銨0.000047%、-15%~+6%。(2)硝酸溶解法平均測得鈉含量、相對誤差分別為高錸酸銨0.000040%、-8%~+5%,錸粉0.000037%、-5%~+5%。(3)硝酸-硫酸溶解法平均測得鈉含量、相對誤差分別為高錸酸0.26mg/L、-12%~+19%,高錸酸銨0.000039%、-27%~+8%,錸粉0.000040%、-22%~+18%;(4)三種方法測得鈉的結果吻合,表明基體錸對鈉的測定影響可忽略不計,水溶解法和硝酸溶解法,操作簡便、快速,且可避免過多操作步驟引入干擾因素。實驗選擇高錸酸用水直接稀釋法、高錸酸銨用熱水溶解法和錸粉用硝酸溶解法。
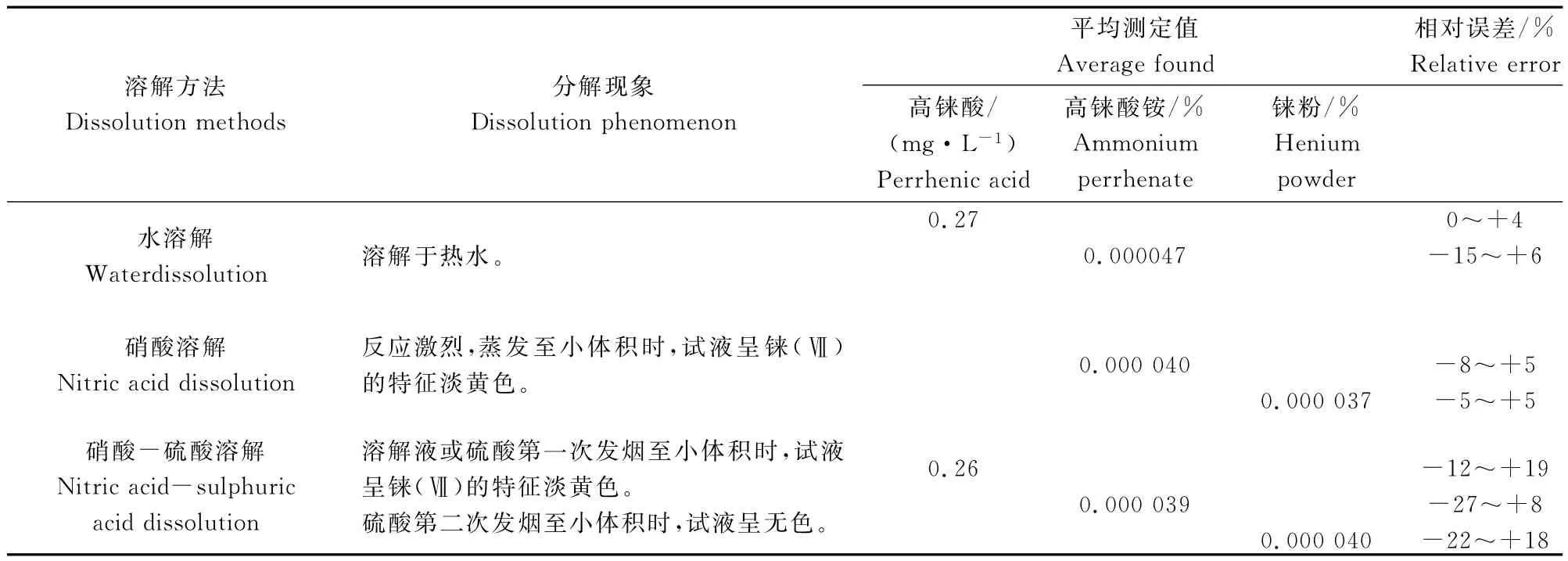
表2 不同溶解方法的比較Table 2 Comparison of analytical results with different dissolution methods
2.2 水溶解、硝酸溶解條件
2.2.1 樣品預處理方式及試劑用量的選擇
分別對高錸酸銨在3~5mL熱水、錸粉在0.4~1.0mL硝酸范圍內進行溶解實驗。結果表明,3~5mL熱水可完全溶解高錸酸銨,但在將試液轉入10mL容量瓶的過程中,用3mL水溶解的試液因冷卻而析出白色鹽類,并沉于杯底,即使用7mL水也不易將其全部轉入容量瓶中,而用4~5mL水溶解的試液,在將試液轉入容量瓶的過程中,即使試液冷卻也無白色鹽類,較易轉入容量瓶中,測得鈉含量、相對誤差為0.000045%~0.000048%、-15%~+6%;0.4~1.0mL硝酸可完全溶解錸粉,但用0.4mL硝酸的溶解反應較慢,用1.0mL硝酸的溶解反應較激烈和試劑空白的吸光度較高,而用0.5mL硝酸的溶解反應較緩和,且試劑空白的吸光度較低,測得鈉含量、相對誤差為0.000037%~0.000041%、-6%~+5%。選擇高錸酸銨加入約5mL水,加熱溶解;錸粉預先加入少量水潤濕,再加入0.5mL硝酸,于室溫溶解至基本無反應,再于低溫加熱溶解,并蒸發至近干,加入約0.5mL水蒸發至近干,反復一次。
2.2.2 樣品量的選擇
分別對高錸酸直接測定或移取5.00mL,稀釋至10.00mL測定的方式進行實驗。結果表明,測得鈉含量、相對誤差分別為直接測定0.26~0.27mg/L、-4%%~+4%,稀釋測定0.27~0.28mg/L、0%~+4%,鑒于鈉標準級差溶液介質為水,實驗選擇高錸酸量為移取5.00mL,稀釋至10.00mL。分別對高錸酸銨、錸粉量在0.5~1.0g范圍內進行實驗。結果表明,用5mL熱水,高錸酸銨約1.0g時,不易被溶解完全;而約0.5g時,易被溶解完全,測得鈉含量、相對誤差為0.000046%~0.000048%、-2%~+2%;錸粉約0.5g時,易被硝酸溶解完全,且試劑空白的吸收光譜較低,測得鈉含量、相對誤差為0.000035%~0.000038%、-5%~+3%。實驗分別選擇高錸酸銨、錸粉量約0.5g。
2.3 測定條件
2.3.1 測定介質的選擇
鈉為堿金屬元素,可于水溶液中穩定存在,分別對二次蒸餾水、超純水中鈉含量進行了測定,結果為二次蒸餾水<0.0005μg/mL、超純水<0.00001μg/mL。實驗選擇超純水為測定介質。
2.3.2 儀器工作條件的選擇
儀器軟件數據庫自帶譜線有靈敏線589.0nm和次靈敏線330.3nm,因樣品中鈉為痕量,選擇靈敏度高的測定譜線589.0nm。
由實驗可知,(1)當乙炔氣流量在0.8~1.5L/min范圍內時,火焰為純藍色、形狀規整,且測得吸光度穩定。(2)燃燒器高度在6.5~7.5mm范圍內時,靈敏度高,且測得吸光度穩定。實驗選擇乙炔氣流量、空氣流量和燃燒器高度分別為1.0L/min、1.0L/min和7.0mm。
2.3.3 濃度線性范圍、線性方程、相關系數及檢出限
根據樣品中鈉含量和測定試液中鈉濃度范圍分別為高錸酸0.27~0.47mg/L和0.135 0~0.235 0μg/mL,高錸酸銨、錸粉0.000040%~0.00049%和0.020 0~0.245 0μg/mL。鈉標準曲線濃度線性范圍0~0.50μg/mL,線性方程y=0.259 7x-0.000 9,吸光度穩定,相關系數R2=0.999 7。分別對水稀釋高錸酸、溶解高錸酸銨,硝酸溶解錸粉的實驗空白溶液進行10次測定,計算出高錸酸、高錸酸銨和錸粉的標準偏差(S)和檢出限(3S)均為1×10-4μg/mL、3×10-4μg/mL。
2.4 方法精密度實驗
移取5.00mL高錸酸于10mL容量瓶中,分別稱取0.5g高錸酸銨、錸粉于50mL燒杯中,按“1.2.1~1.2.2”進行分析,以計算方法的精密度。由表3可知,測定高錸酸0.27~0.47mg/L,高錸酸銨、錸粉0.000040%~0.00049%的鈉含量,極差、標準偏差(S)和相對標準偏差(RSD)分別為高錸酸±0.07mg/L、0.027%和6%~10%,高錸酸銨、錸粉±0.000013%~0.00015%、0.000002%~0.00004%和5%~9%。
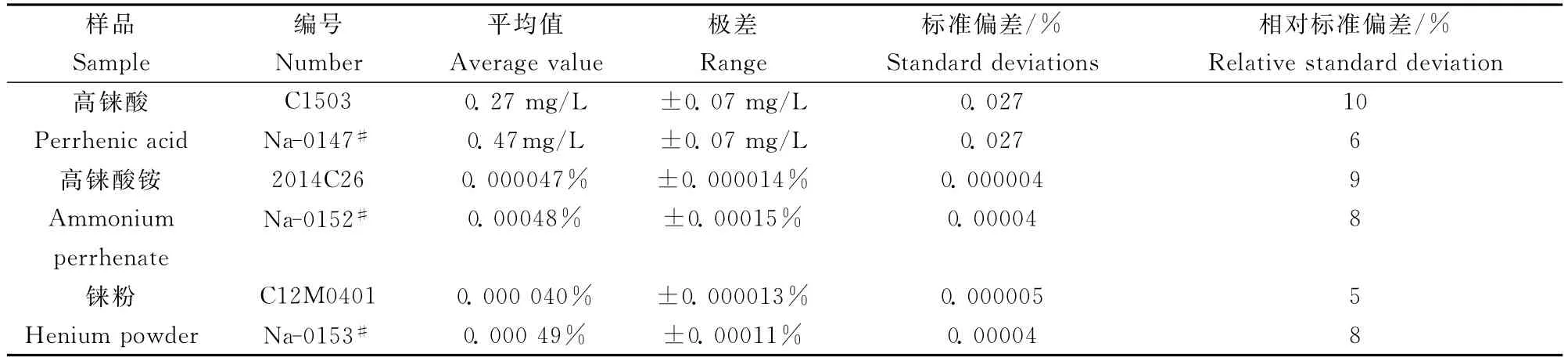
表3 樣品分析結果Table 3 Analytical results of sodium in samples(n=7)
2.5 準確度實驗
于含0.023 5~0.245 0μg/mL的鈉試液中,加入0.050 0~0.2000μg/mL的鈉標準溶液,測定鈉加標回收率,以計算方法的準確度。由表4可知,測得鈉標準量和加標回收率分別為高錸酸0.048 8~0.100 8μg/mL和97.6%~101.6%,高錸酸銨、錸粉0.050 0~0.202 3μg/mL和96.2%~103.1%。
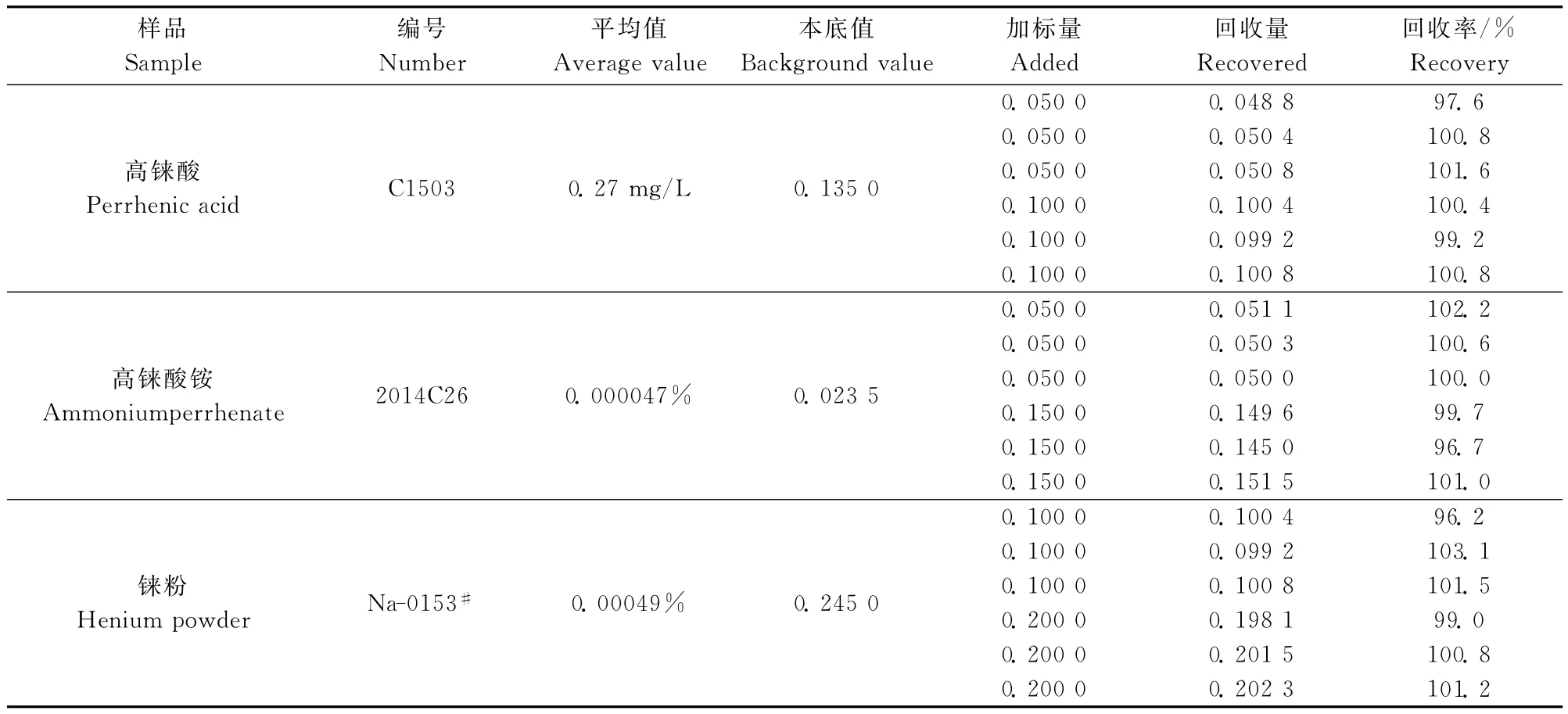
表4 樣品加標回收率Table 4 Recovery tests of the method /(μg·mL-1)
3 結語
水溶解法、硝酸溶解法或硝酸-硫酸溶解法分解樣品完全,鈉的分析結果吻合,但前者操作較簡便;火焰原子吸收法測定高錸酸、高錸酸銨和錸粉中鈉含量,分析結果準確、精密,滿足錸產品質量控制對鈉分析誤差的要求。
[1]董海剛,劉楊,范興祥,等 .錸的回收技術研究進展[J].有色金屬(冶煉部分)[NonferrousMetals(Extractive Metallurgy)],2013(6):30-33.
[2]李紅梅,賀小塘,趙雨,等 .錸的資源、應用和提取[J].貴金屬(PreciousMetals),2014,35(2):77-81.
[3]李建,計輝,錢玉萍 .電感耦合等離子體發射光譜(ICPOES)法同時測定活性炭中鋁、鈷、鎘、銅、鐵、鎂、錳、鈉、磷、硫10種元素[J].中國無機分析化學(ChineseJournal ofInorganicAnalyticalChemistry),2012,2(1):58-60.
[4]樊穎果,徐國津 .原子吸收光譜和原子發射光譜法測定酸雨中鉀、鈉、鈣、鎂方法比較[J].中國無機分析化學(ChineseJournalofInorganicAnalyticalChemistry),2013,3(2):28-31.
[5]孫建之,鄧小川,宋士濤 .離子色譜法測定高純氯化鋰中的微量雜質離子[J].海湖鹽與化工(JournalofSalt andChemicalIndustry),2005,34(3):24-26.
[6]任慧,孫建新.AAS法測定純鉿中痕量元素鈉[J].分析儀器(AnalyticInstrument),2014(4):64-67.
[7]王皓瑩 .火試金-原子吸收光譜法測定錫陽極泥中金、銀含量[J].中國無機分析化學(ChineseJournalofInorganicAnalyticalChemistry),2015,5(2):59-61.
[8]陳雯,金婭秋,梁潔,等 .原子吸收法測定三氯化釕化合物中鈉[J].貴金屬(PreciousMetals),2013,34(1):70-73.
[9]全國有色金屬標準化委員會.YS/T 594—2006硝酸銠[S].北京:中國標準出版社,2006.
[10]楊貴生,楊文生 .原子吸收光譜法測定加氫催化劑中的鈉[J].當代化工(ContemporaryChemical Industary),2007,36(1):92-93.
[11]郭端陽,王克勤,李愛秀,等 .火焰原子吸收光譜法測定拜耳法生產氧化鋁赤泥中鈉[J].冶金分析(MetallurgicalAnalysis),2012,32(2):52-55.
[12]肖忠峰,趙西梅,于祥,等 .火焰原子吸收光譜法測定土壤及植物中鈉含量[J].化學工程師(ChemicalEngineer),2012,48(12):1444-1449.
[13]劉波 .火焰原子吸收光譜法測定食用油脂中鈉含量[J].理化檢驗-化學分冊[Ptca(PartB:Chem.Anal.)],2012,48(12):1444-1449.
[14]全國有色金屬標準化委員會.YS/T 836—2012高錸酸[S].北京:中國標準出版社,2012.
[15]全國有色金屬標準化委員會.YS/T 833—2012錸酸銨化學分析方法 錸酸銨中鈹、鎂、鋁、鉀、鈣、鈦、鉻、錳、鐵、鈷、銅、鋅和鉬量的測定 電感耦合等離子體原子發射光譜法[S].北京:中國標準出版社,2012.
Determination of Trace Sodium in Rhenium Products by Flame Atomic Absorption Spectrometry
TANG Hui1,SHU Bo1,ZHOU Dongmei1,ZHANG Xuandong2,HUA Hongquan1,YANG Kunbin1,SHI Yifeng1,DAI Hongkun1,WANG Chuanfei1,ZHENG Wenying1,LI Jun1,FANG Yong1,WU Zhiqing1,LIN Bo2,LIU Wen2,ZHU Liya2*
(1.YunnanCopperCo.Ltd.,Kunming,Yunnan650102,China;2.StateKeyLaboratoryofAdvancedTechnologiesforComprehensive UtilizationofPlatinumMetals,SinoPlatinumMetalsResources(Yimen)Co.,Ltd,Kunming,Yunnan650106,China)
A method for the determination of trace sodium in perrhenic acid,ammonium perrhenate and rhenium powder by flame atomic absorption spectrometry was proposed.Samples pretreatment and experimental conditions for sodium analysis were researched.The results indicated that analytical results by water dissolving method were in good accord with those obtained by nitric acid dissolution method and nitric acid-sulphuric acid dissolution methods.Sodium concentration had a good linear relationship in the range of 0.020 0~0.500 0μg/mL under the optimized conditions.The concentration ranges for perrhenic acid,ammonium perrhenate and rhenium powde were 0.27~0.47mg/L,0.000047%~0.00048%and 0.000040%~0.00049%,respectively.The detection limits,relative standard deviations(RSDs,n=7)and recoveries for sodium were 3×10-4μg/mL,6%~10%and 98%~102%(perrhenic acid),3×10-4μg/mL,8%~9%and 96%~102%(ammonium perrhenate)and 3×10-4μg/mL,5%~9%and 96%~103%(rhenium powde),respectively.The proposed method is accurate,rapid and convenient,and has been applied to the determination of trace sodium in actual sample with satisfactory results.
sodium;trace;rhenium products;flame atomic absorption spectrometry
O657.31;TH744.12
A
2095-1035(2015)04-0060-05
2015-08-13
2015-09-08
云南銅業股份有限公司基金項目(2014JK003、2014JK005、2014JK006)、貴研資源(易門)有限公司橫向合作基金項目(GYZY-2014-001)資助
唐慧,女,工程師,主要從事有色金屬分析研究與應用。E-mail:tanghui@163.com
*通信作者:朱利亞,女,教授,主要從事稀貴金屬分析和微波消解研究與應用。E-mail:zzly200686@163.com
10.3969/j.issn.2095-1035.2015.04.014
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
