16S核糖體DNA宏基因組測序中細菌核酸提取方法的比較研究
2015-05-04 08:03:50王偉王玉琢舒鵬米志強安小平裴廣倩劉文麗袁文俊史套興童貽剛
生物技術通訊 2015年4期
關鍵詞:方法
王偉,王玉琢,舒鵬,米志強,安小平,裴廣倩,劉文麗,袁文俊,史套興,童貽剛
1.安徽醫科大學,安徽 合肥 230032;2.軍事醫學科學院 微生物流行病研究所,北京 100071;
3.寧夏醫科大學,寧夏 銀川 750000
人和動物胃腸道內存在大量微生物,它們與宿主的生理功能密切相關,這些微生物群落之間及微生物與動物宿主之間形成了相互依存、相互作用的不可分割的整體。傳統的微生物學研究方法主要是對微生物進行培養和分離。到目前為止,絕大多數微生物(99%以上)無法依靠培養的方式獲得[1],這極大地限制了人們對微生物的研究。
近年來,隨著高通量測序技術的應用[2-3],宏基因組學發展迅速[4],為我們了解環境和人體自身微生物種群提供了新的途徑和視野。宏基因組學與傳統微生物研究方式的最大區別在于把微生物看成一個整體,擺脫了對單個微生物的培養和分離步驟,直接對環境中所有的微生物進行研究,進而可以全面地對所有微生物進行分析。隨著宏基因組學的興起和發展,人們對人體的腸道、口腔、皮膚組織等中的微生物種群進行了研究[5],發現了多種新的細菌,了解了這些細菌和人體健康之間的關系。
然而,從最初的細菌核酸提取到后期的數據分析,仍然有較多技術性和科學性問題制約著宏基因組學特別是16S核糖體DNA(ribosomal DNA,rDNA)宏基因組學的發展。如細菌核酸的提取[6]、擴增高變區的選擇、通用引物的選擇[7]、測序平臺的選擇以及后期數據分析策略的選擇,都會對宏基因組測序的結果產生影響。對于來自環境中的復雜樣本,細菌核酸提取的效率和質量至關重要,是后續擴增、測序和分析的基礎。目前細菌核酸提取的方法有很多種,原理和操作步驟各不相同,各有優缺點[8-10]。在此,我們采用TRIzol法、試劑盒法,以及在試劑盒法基礎上進行5種改進,共7種方法對模擬樣本進行核酸提取,并特異性地擴增16S rDNA的V1~V2高變區,之后構建測序文庫,分析測序結果中16S rDNA的分布,還原模擬樣本中各種細菌的含量和相對分布,尋找一種廣譜、高效的細菌核酸提取方法。
1 材料和方法
1.1 材料
選取10種在微生物學與分子生物學特征上具有顯著差異的細菌,包括革蘭陽性和陰性菌,分別是金黃色葡萄球菌(Staphylococcus aureus)、弗氏檸檬酸桿菌(Citrobacter freundii)、肺炎克雷伯菌(Klebsi?ella pneumoniae)、奇異變形桿菌(Proteus mirabilis)、腸道沙門菌(Salmonella enterica)、蠟樣芽孢桿菌(Bacillus cereus)、摩氏摩根菌(Morganella morga?nii)、海藻希瓦菌(Shewanella algae)、宋內志賀菌(Shigella sonei)、粘質沙雷菌(Serratia marcescens)。以上菌種均分離自解放軍307醫院檢驗科。
TRIzol(Thermo Fisher Scientific);Roche High PurePCR TemplatePreparationKitVersion 2.0(Roche);溶葡萄球菌酶、溶菌酶(Sigma);DNA Marker(北京全式金公司);Q5 High-Fidelity 2×Master Mix、Fast DNA Library Prep Set for Ion Torrent(New England Biolabs);QIAquick Gel Ex?traction Kit(QIGEN)。
PCR 儀(Gene Amp PCR System 9700);Ion torrent測序儀(Thermo Fisher Scientific);NanoDrop system;超聲波儀(南京新辰生物公司)。
1.2 細菌培養
將上述菌株分別接種于LB固體培養基上,37℃培養18 h,挑取單克隆菌落接種于LB液體培養基中,37℃、150 r/min振蕩培養至對數后期或穩定期后于4℃保存。
1.3 菌種鑒定
分別取以上經液體培養基培養后的菌液500 μL于1.5 mL離心管中,沸水浴10 min后作為16S rDNA PCR鑒定模板。上游引物為27F(5'-AGAGT TTGATCCTGGCTCAG-3'),下游引物為 1492R(5'-TACGACTTAACCCCAATCGC-3'),產 物 1.4kb。PCR 體系:上、下游引物各2 μL,Q5 High-Fidelity 2× Master Mix 25 μL,DNA 模板 2 μL,用無菌ddH2O補足50 μL。PCR條件:預變性95℃ 5 min;變性95℃ 30 s,退火55℃ 30 s,延伸72℃ 1 min(30個循環);延伸7 min。PCR擴增產物經3730測序,進行菌種鑒定。
1.4 細菌鋪板計數和等比例混合
將菌液稀釋至1/106、1/107、1/108共3個梯度后進行涂布平板計數,根據細菌數將10種菌等量混合于1.5 mL EP管中,12 000 r/min離心3 min,棄上清,用PBS洗滌2次后收集細菌。
1.5 細菌DNA的提取
1.5.1 TRIzol提 取 向 混 合 細 菌 中 加 入1 mL TRIzol后用移液器反復吹打,室溫靜置15 min,加入0.2 mL氯仿充分振蕩混勻,室溫靜置3 min,4℃、10 000 r/min離心10 min,棄上層水相,保留中間相與下層有機相,向中間相及有機相加入0.3 mL無水乙醇,上下顛倒混勻,室溫放置30 min,4℃、2000 r/min離心5 min,棄上清,用1 mL 0.1 mol/L檸檬酸鈉溶液(10%乙醇)洗滌沉淀,室溫放置30 min,期間振蕩數次,4℃、2000 r/min離心5 min,棄上清,用1.5 mL 75%乙醇洗滌DNA沉淀,室溫放置30 min,期間振蕩數次,4℃、2000 r/min離心5 min,棄上清后充分晾干DNA沉淀,加入50 μL 8 mmol/L NaOH溶液溶解DNA沉淀,-20℃保存。
1.5.2 試劑盒直接提取 按照Roche High Pure PCR Template Preparation Kit Version 2.0 中 Isola?tion of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.5.3 溶葡酶處理后試劑盒提取 向混合細菌中加入溶葡酶(終濃度 1 mg/mL)20 μL,按照 Roche High Pure PCR Template Preparation Kit Version 2.0中Isolation of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.5.4 超聲波處理后試劑盒提取 將混合細菌置于冰水浴中,間歇超聲波處理(超聲2 s,間隙5 s,功率20%,總時 5 min),按照 Roche High Pure PCR TemplatePreparation KitVersion 2.0 中 Isolation of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.5.5 超聲波處理并加溶葡酶處理后試劑盒提取
將混合細菌置于冰水浴中,間歇超聲波處理(超聲2 s,間隙5 s,功率20%,總時5 min),加溶葡酶(終濃度 1 mg/mL)20 μL,按照Roche High Pure PCR TemplatePreparation KitVersion 2.0 中 Isolation of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.5.6 勻漿處理后試劑盒提取 將混合細菌勻漿處理(30 Hz,5 min),按照 Roche High Pure PCR TemplatePreparation KitVersion 2.0 中 Isolation of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.5.7 勻漿處理并加溶葡酶處理后試劑盒提取 將混合細菌勻漿處理(30 Hz,5 min),加入溶葡酶(終濃度 1 mg/mL)20 μL,按照Roche High Pure PCR TemplatePreparation KitVersion 2.0 中 Isolation of Nucleic Acids form Bacteria方案提取細菌DNA,于-20℃保存。
1.6 細菌基因組DNA分析
提取后的產物用NanoDrop system檢測DNA的濃度和純度,1%瓊脂糖凝膠電泳分析混合細菌基因組DNA的完整性。
1.7 擴增細菌16S rDNA的V1~V2高變區
用通用引物[11]擴增細菌16S rDNA的V1、V2高變區,上游引物為27F(5'-AGRGTTYGATYMTGGC TCAG-3'),下游引物為338R(5'-TGCTGCCTCCCG TAGGAGT-3')。PCR體系:上、下游引物各2 μL,Q5 High-Fidelity 2× Master Mix 25 μL,DNA模板2 μL,用無菌ddH2O補至50 μL。PCR條件:預變性95℃ 5 min;變性 95℃ 30 s,退火 55℃ 30 s,延伸72℃ 30 s(30個循環);延伸7 min。2%瓊脂糖凝膠電泳回收目的條帶,用QIAquick Gel Extraction Kit純化。
1.8 測序文庫的構建及高通量測序
投入100 ng擴增產物,參照Fast DNA Library Prep Set for Ion Torrent說明書,構建高通量測序文庫約400 bp,之后進行PGM測序。
1.9 生物信息學分析
用 NGS QC Toolkit v2.3.3(參 數 :-l 70-s 20-n 8-m 100-c 8)[12]去除低質量及低復雜度序列,依據樣品前加的標簽序列,用python腳本將7種方法的數據進行分割提取,再用CLC Genomics Workbench v3.6.1(丹麥 CLC Inc,http://www.clcbio.com)將數據比對前期細菌鑒定的16S rDNA全長序列,用MEGA(v.6.0.5)軟件[13]構建細菌16S rDNA進化樹。
2 結果
2.1 混合樣本的細菌組成
本次實驗共挑選了10種細菌,這10種菌在微生物學與分子生物學特征上具有顯著差異,包括革蘭陽性菌和革蘭陰性菌。細菌基因組16S rDNA拷貝數為 5~14,16S rDNA 鑒定長度為 1439~1470 bp,16S rDNA GC 含 量 為 51.00% ~55.20% ,擴 增 的V1~V2高變區長度為310 bp,V1~V2的GC含量為51.3%~57.6%,通用引物與細菌16S rDNA的匹配性較好(表1)。
從細菌16S rDNA進化樹(圖1)可以看出10種菌的進化距離較遠,差異較為明顯,有利于后續測序數據不同細菌的分類。
2.2 不同方法提取的細菌DNA含量與純度分析
用不同提取方法得到的細菌DNA含量與純度不同(表2),TRIzol法提取的細菌DNA含量最高,勻漿處理后試劑盒提取的細菌DNA含量次之;超聲波處理后試劑盒提取和勻漿加溶葡酶處理后試劑盒提取的細菌DNA純度最高,超聲波加溶葡酶后試劑盒提取的次之。
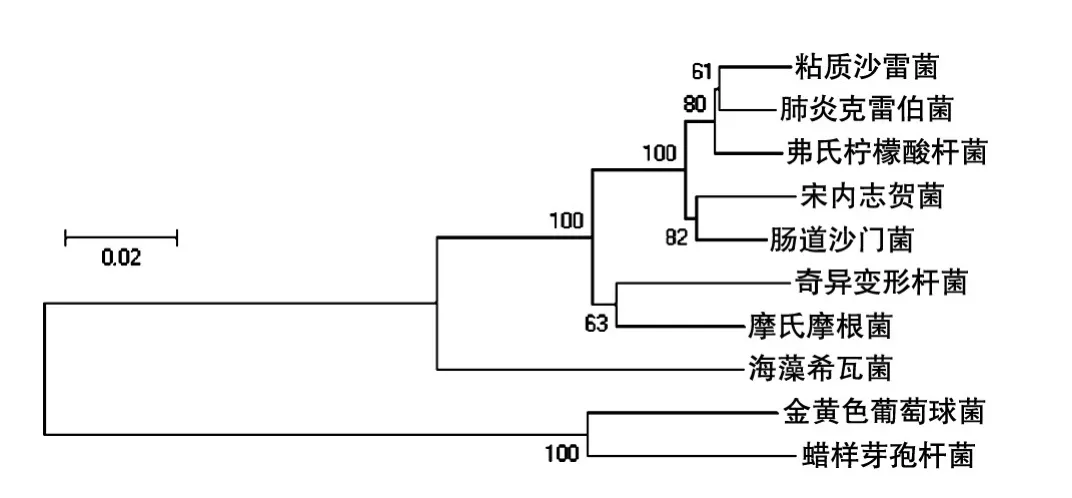
圖1 實驗所用細菌16S rDNA進化樹
2.3 不同方法提取的細菌DNA完整性分析
2%瓊脂糖凝膠電泳分析表明,試劑盒提取及超聲波結合試劑盒提取的模擬樣品細菌DNA具有較好的完整性(圖2);TRIzol提取、溶葡酶加試劑盒提取、超聲波加溶葡酶加試劑盒提取、勻漿加試劑盒提取及勻漿加溶葡酶加試劑盒提取的模擬樣品細菌DNA分子出現了顯著的彌散,表明DNA分子產生了斷裂,完整性差。
2.4 不同方法的高通量測序結果分析
盡管7種方法中10種細菌是等比例等量投入的,但測序結果差別很大(表3),在TRIzol法和超聲波加試劑盒法數據中,前三位細菌相同,分別為蠟樣芽孢桿菌、肺炎克雷伯菌和弗氏檸檬酸桿菌;在TRIzol法、試劑盒法、超聲波加試劑盒法和勻漿加試劑盒法數據中,含量最低的3種菌為海藻希瓦菌、奇異變形桿菌和金黃色葡萄球菌;而在溶葡酶加試劑盒法、超聲波加溶葡酶加試劑盒、勻漿加溶葡酶加試劑盒法中,含量最低的2種菌也是海藻希瓦菌和奇異變形桿菌。在溶葡酶加試劑盒法、超聲波加溶葡酶加試劑盒法、勻漿加溶葡酶加試劑盒法數據中,金黃色葡萄球菌的數據量明顯高于其他4種提取法。蠟樣芽孢桿菌、金黃色葡萄球菌和奇異變形桿菌基因組16S rDNA拷貝數分別為14、5、5,在TRIzol法、超聲波加試劑盒法、勻漿加試劑盒法數據中,蠟樣芽孢桿菌數據比重要高于金黃色葡萄球菌和奇異變形桿菌,表明細菌16S rDNA拷貝數對于宏基因組檢測有影響。弗氏檸檬酸桿菌和腸道沙門菌基因組16S rDNA拷貝數都為9次,通用引物27F與弗氏檸檬酸桿菌和腸道沙門菌的匹配性分別為20/0、16/4,在勻漿加溶葡酶加試劑盒法數據中,弗氏檸檬酸桿菌的數據比重高于腸道沙門菌,表明通用引物與細菌16S rDNA匹配性對于宏基因組檢測有影響。
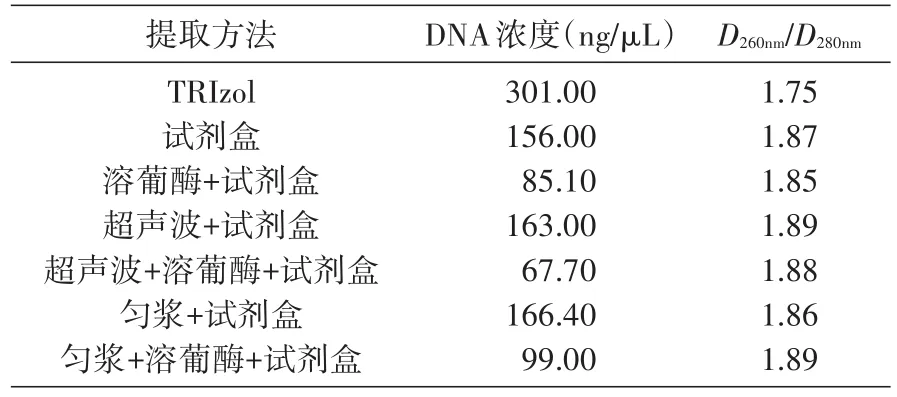
表2 不同提取方法所得細菌DNA含量和純度測定結果
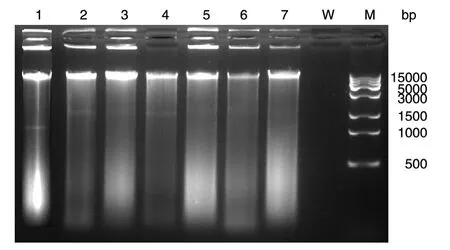
圖2 不同提取方法和前處理方法提取細菌DNA電泳分析1:TRIzol提取產物;2:試劑盒提取產物;3:溶葡酶加試劑盒提取產物;4:超聲波加試劑盒提取產物;5:超聲波加溶葡酶加試劑盒提取產物;6:勻漿加試劑盒提取產物;7:勻漿加溶葡酶加試劑盒提取產物;W:水;M:DNA marker
對7組數據中10種菌的相對分布進行統計和計算,繪制柱形圖(圖3),可以直觀地觀察到超聲波加試劑盒提取方法中各種菌的數據分布相對均勻。
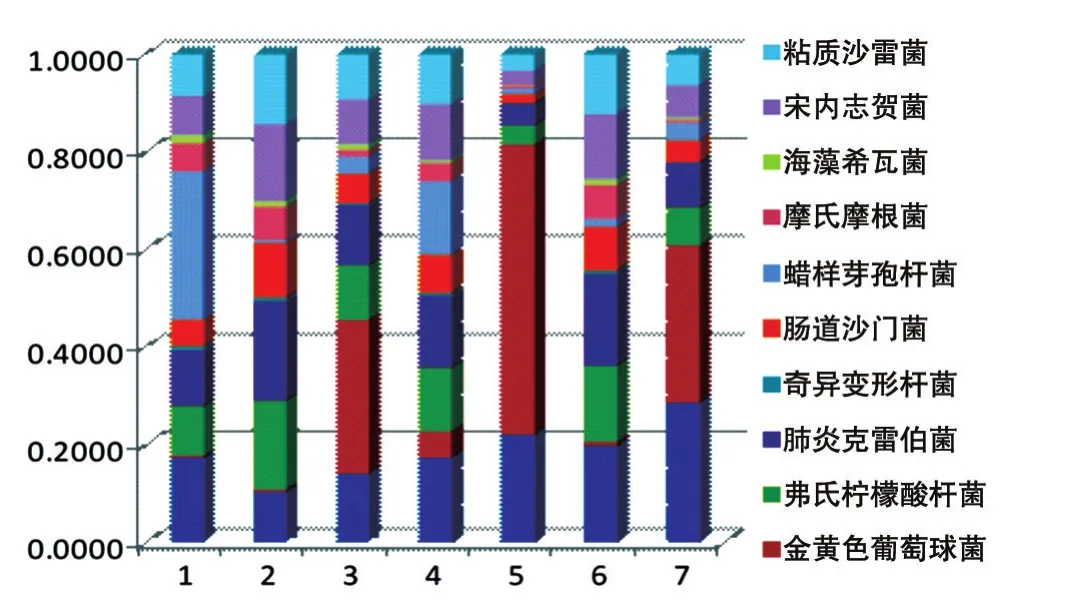
圖3 不同方法中各種細菌數據量的相對分布
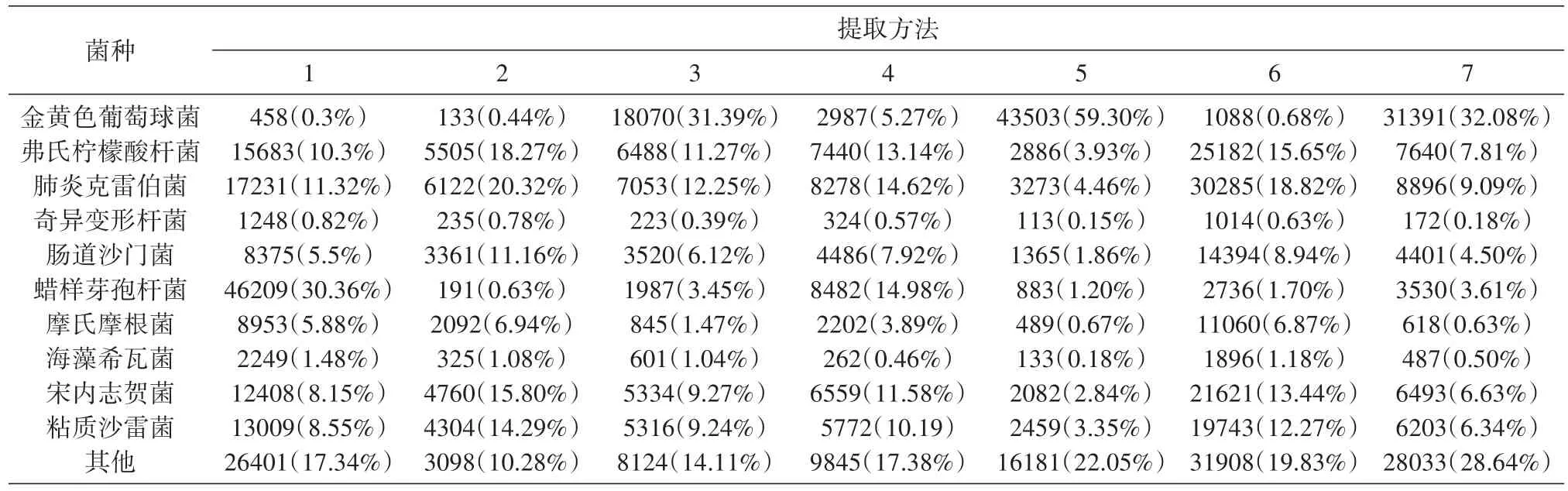
表3 不同方法測序結果中比對上不同細菌的序列條數分布
3 討論
目前,基于16S rDNA測序的宏基因組學發展迅速。盡管此技術在探索各種環境樣本中細菌物種的多樣性方面具有無可比擬的優勢,但也存在技術上的不足和缺陷,如在樣本的收集和保存、核酸的提取、擴增使用的引物、測序平臺的選擇等方面的不足,進而直接導致實驗結果不能真實地反映細菌物種的多樣性。本次實驗在排除其他影響因素的情況下,探究了細菌DNA提取方法對于16S rDNA測序的影響。結果表明,不同的前處理及提取方法對于細菌DNA的提取影響顯著,在同一方法中,不同細菌的提取效果差別很大,對同一種細菌使用不同方法提取的效果差別也很大。在加入溶葡酶的3種方法中,金黃色葡萄球菌的數據量遠高于另外4種不加溶葡酶的方法,說明溶葡酶處理對于樣本中金黃色葡萄球菌的檢測至關重要。同時實驗結果顯示細菌16S rDNA拷貝數及通用引物與細菌16S rDNA的匹配性對于宏基因組檢測也有影響。在超聲波加試劑盒提取測序數據中,各種細菌的數據量分布相對均勻,表明對各種細菌的提取效果較好,亦即超聲波處理能夠改善試劑盒的提取效果。在其他幾種提取方法中,各種細菌的數據量差別很大,無明顯的規律可循。
對于來自環境中的真實樣本,其復雜度遠遠高于本實驗所用的模擬樣本。真實樣本往往含有更多種類的細菌、不同的化學組分,以及來自其他有機體的細胞和DNA成分,在宏基因組學研究中應當考慮到這些因素。此次實驗探索尋找廣譜高效的細菌DNA提取方法,提高了宏基因組學測序技術反映真實環境樣品中物種多樣性的能力。
[1] Kaeberlein T,Lewis K,Epstein S S.Isolating"uncultivable"microorganisms in pure culture in a simulated natural environ?ment[J].Science,2002,296(5570):1127-1129.
[2] Mardis E R.Next-generation DNA sequencing methods[J].An?nu Rev Genomics Hum Genet,2008,9:387-402.
[3] von Bubnoff A.Next-generation sequencing:the race is on[J].Cell,2008,132(5):721-723.
[4] Handelsman J,Rondon M R,Brady S F,et al.Molecular bio?logical access to the chemistry of unknown soil microbes:a new frontier for natural products[J].Chem Biol,1998,5(10):R245-R249.
[5] Qin J,Li R,Raes J,et al.A human gut microbial gene cata?logue established by metagenomic sequencing[J].Nature,2010,464(7285):59-65.
[6] Salonen A,Nikkil J,Jalanka-Tuovinen J,et al.Comparative analysis of fecal DNA extraction methods with phylogenetic microarray:effective recovery of bacterial and archaeal DNA using mechanical cell lysis[J].J Microbiol Methods,2010,81(2):127-134.
[7] Hong S,Bunge J,Leslin C,et al.Polymerase chain reaction primersmisshalfofrRNA microbialdiversity[J].ISME J,2009,3(12):1365-1373.
[8] Zhou J,Bruns M A,Tiedje J M.DNA recovery from soils of diverse composition[J].ApplEnviron Microbiol,1996,62(2):316-322.
[9] Lffler J,Hebart H,Schumacher U,et al.Comparison of dif?ferentmethodsforextraction ofDNA offungalpathogens from cultures and blood[J].J Clin Microbiol,1997,35(12):3311-3312.
[10]Yen T,Six D L,Burke E J.Evaluation of rapid DNA extrac?tion and identification of Phellinus pini associated with Pinus contorta by rDNA assay[J].Forest Products J,2006,56(11/12):107.
[11]Wu G D,Lewis J D,Hoffmann C,et al.Sampling and pyro?sequencing methodsforcharacterizingbacterialcommunities in the human gut using 16S sequence tags[J].BMC Microbi?ol,2010,10(1):206.
[12]Patel R K,Jain M.NGS QC Toolkit:a toolkit for quality control of next generation sequencing data[J].PloS One,2012,7(2):e30619.
[13]Tamura K,Stecher G,Peterson D,et al.MEGA6:molecular evolutionary genetics analysis version 6.0[J].Mol Biol Evol,2013,30(12):2725-2729.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
