敗醬皂苷對照品制備工藝研究△
2015-09-25 12:59:16陸昊劉宏尉王增尚劉江云郝麗莉楊世林
中國現代中藥 2015年6期
陸昊,劉宏尉,王增尚,劉江云,郝麗莉,楊世林
(蘇州大學 藥學院,江蘇 蘇州 215123)
·中藥工業·
敗醬皂苷對照品制備工藝研究△
陸昊,劉宏尉,王增尚,劉江云*,郝麗莉,楊世林
(蘇州大學 藥學院,江蘇 蘇州 215123)
目的:對黃花敗醬總皂苷進行分離,以獲得高純度的3種敗醬皂苷對照品。方法:綜合采用多種硅膠、反相硅膠、凝膠柱色譜和中壓、高壓制備色譜技術,對敗醬總皂苷進行分離純化,采用HPLC-ELSD檢測樣品含量。結果:制備獲得的化合物1~3峰純度均為98.9%以上。結論:該制備工藝簡潔、高效,可獲得高純度的敗醬皂苷對照品。
敗醬;皂苷;對照品;分離
敗醬是傳統中藥,始載于《神農本草經》,列為中品,其性微寒,味苦、辛,歸肺、大腸、肝經。具有清熱解毒,排膿破瘀之功能,可用于治療腸癰、下痢、赤白帶下、產后瘀滯腹痛、目赤腫痛、癰腫疥癬等癥。現代研究證明敗醬具有鎮靜催眠、抗菌、抗病毒、抗腫瘤、保肝利膽等多種藥理作用,臨床上可用于治療神經衰弱、肝炎、肝硬化、流行性腮腺炎、鼻竇炎和結腸炎等[1]。
本課題組對黃花敗醬中的化學成分進行系統分離,鑒定了主要的17種三萜皂苷及其他化合物[2-4]。進一步研究及文獻[5]分析結果表明,齊墩果酸型皂苷具有較好的抗腫瘤活性。本文報道敗醬總皂苷中3種主要成分對照品的制備工藝,為黃花敗醬的后續研究與開發提供參考。
1 儀器、材料與試劑
1.1 儀器
高效液相色譜系統(LC-20AD輸液泵、SPD-M20A紫外檢測器、CTO-20A柱溫箱、LC-Solution色譜工作站,日本島津公司);ODS色譜柱(PAQ-C18,日本Cosmosil公司);BP-Purifier-100中壓液相系統(蘇州利穗有限公司);HB-DAC-100動態軸向制備柱系統(江蘇漢邦科技有限公司);Agilent-6100系列LCMS系統;Avance III plus 400 MHz核磁共振儀(德國Bruker Daltonics)。
1.2 材料
黃花敗醬的全草(云南蒙自縣,批次:PS121010)于2012年6~8月間采集,經蘇州大學陸
葉博士鑒定為敗醬科敗醬屬植物黃花敗醬PatriniascabiosaefoliaFisch.的全草。
1.3 試劑
大孔吸附樹脂LX60(西安藍曉科技有限公司);硅膠300~400目(青島海洋化工有限公司);反相C18硅膠(75 μm,日本Cosmosil公司);Sephadex LH-20凝膠(美國GE公司);甲醇(色譜純,上海化學試劑有限公司);純凈水(杭州娃哈哈公司);去離子水(自制);其他試劑均為分析純。
2 方法
2.1 液相分析條件
采用高效液相-蒸發光散射檢測(HPLC-ELSD)分析方法,液相分析條件:蒸發光散射檢測器的漂移管溫度為40 ℃;霧化氣壓力為350 kPa;氮氣流速為2.5 L·min-1;流動相為甲醇(A)-0.5%甲酸水溶液(B),梯度洗脫(0~30 min,70%→100%A);柱溫為35 ℃;流速為1 mL·min-1;進樣量為20 μL。
2.2 黃花敗醬總皂苷的制備
筆者根據抗腫瘤藥理實驗結果,研究建立了總皂苷精制工藝:取黃花敗醬全草20 kg,用10倍量75%乙醇回流提取兩次,每次2 h,合并提取液,濃縮,加水至40 L,用氫氧化鈉(NaOH)調節pH至11~13之間,保持微沸水解4 h,靜置放冷,以氯化氫(HCl)調節pH至10,加水至40 L,上大孔吸附樹脂(20 L·BV-1),以水(4 BV)、10%乙醇(4 BV)、80%乙醇(6 BV)洗脫;收集80%乙醇洗脫部位,濃縮,干燥,即得敗醬總皂苷(170 g)。見圖1。
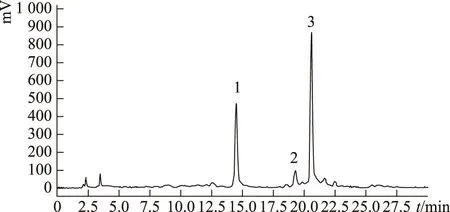
注:1~3.敗醬皂苷,結構式參見圖2。圖1 敗醬總皂苷HPLC圖
3 結果
3.1 分離純化
稱取敗醬總皂苷20 g,經減壓硅膠柱柱色譜,以三氯甲烷-甲醇(10∶1→5∶5)系統溶劑梯度洗脫,TLC檢測,合并相同組分,獲得流分F1~F16。
取F9(4.23 g)經硅膠H柱分離,氯仿-甲醇(CHCl3-CH3OH)(9∶1→8∶2)梯度洗脫,獲得F9.1~F9.6,其中F9.3上中壓反相碳18硅膠(ODS)柱,以CH3OH-H2O 40∶60、60∶40、65∶35、100∶0分段洗脫,其中65%洗脫流分經高效液相色譜法(HPLC)檢測合并,濃縮后經Sephadex LH-20,以CHCl3-CH3OH(1∶1)純化,獲得化合物1(0.45 g),按液相相對峰面積計純度為99.1%。
取F12(8.16 g)經硅膠H柱分離,CHCl3-CH3OH(7∶3→5∶5)梯度洗脫,獲得F12.1~F12.9,合并F12.5~F12.7上中壓ODS柱,以CH3OH-H2O 40∶60、75∶25、100∶0分段洗脫,其中75%流分合并,進一步經反相動態軸向柱色譜以CH3OH-H2O按70∶30(F12.5.1~F12.5.4)、75∶25(F12.5.5~F12.5.15)、100∶0(F12.5.16~F12.5.20)進行分離,其中流分F12.5.8~F12.5.11、F12.5.13~F12.5.14分別經HPLC檢測合并,經Sephadex LH-20,以CHCl3-CH3OH(1∶1)純化,依次獲得化合物2(82 mg)、3(0.95 g),按液相相對峰面積計純度依次為98.9%、99.2%。
3.2 結構鑒定
筆者采用HR-ESI-MS、1H-NMR、13C-NMR等譜學技術,參考相關文獻,對化合物1~3進行結構鑒定,各化合物結構式見圖2。
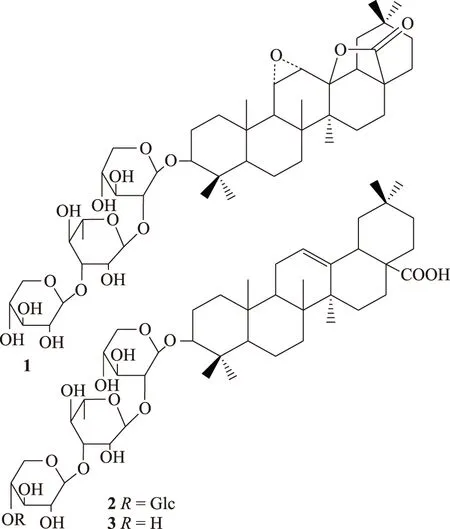
圖2 敗醬皂苷1~3結構式
化合物1:白色無定形粉末(甲醇)。HR-ESI-MS給出[M-H]-準分子離子m/z881.480 2(計算值為881.484 5),相應分子式為C46H72O16。1H-NMR(400 MHz,C5D5N)和13C-NMR(100 MHz,C5D5N)數據與文獻[4]報道的3-O-β-D-吡喃木糖基-(1→3)-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃木糖基-11α,12α-環氧齊墩果烷-28,13β-交酯一致。
化合物2:白色粉末(甲醇),在TLC板上用10%硫酸-乙醇顯色為紫紅色,HR-ESI-MS給出[M-H]-準分子離子m/z 1 028.552 5(計算值為1 028.56),相應分子式為C52H84O20。1H-NMR(400 MHz,C5D5N)δ:6.38(1H,br s,H-1 of Rha),5.26(1H,d,J=8.0 Hz,H-1′ of Xyl′),5.12(1H,d,J=8.0 Hz,H-1 of Glc),4.86(1H,d,J=7.0 Hz,H-1 of Xyl),5.26(1H,br s,H-12),1.42(3H,d,J=6.0 Hz,CH3-6 of Rha),1.15(3H,s,CH3-23),1.10(3H,s,CH3-27),1.05(3H,s,CH3-24),1.01(3H,s,CH3-26),0.81(3H×3,s,CH3-29,30,25);13C-NMR(100 MHz,C5D5N)數據見表1。以上數據與文獻[3,6]報道的3-O-β-D-吡喃葡萄糖基-(1→4)-β-D-吡喃木糖基-(1→3)-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃木糖基-齊墩果酸-28-O-β-D-吡喃葡萄糖苷比較,為該化合物經堿水解脫去28-葡萄糖基的次生皂苷,鑒定其為齊墩果酸-3-O-β-D-吡喃葡萄糖基-(1→4)-β-D-吡喃木糖基-(1→3)-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃木糖苷。
化合物3:白色粉末(甲醇),HR-ESI-MS給出[M-H]-準分子離子m/z 865.497 1(計算值為865.498 0),相應分子式為C46H74O15。1H-NMR(400 MHz,C5D5N)δ:6.59(1H,s,H-1 of Rha),5.45(1H,d,J=7.5 Hz,H-1′ of Xyl′),5.01(1H,d,J=7.0 Hz,H-1 of Xyl),5.39(1H,br s,H-12),1.63(3H,d,J=6.0 Hz,CH3-6 of Rha),1.39(3H,s,CH3-23),1.30(3H,s,CH3-27),1.21(3H,s,CH3-24),0.99(3H,s,CH3-26),0.96(3H,s,CH3-29),0.94(3H,s,CH3-30),0.82(3H,s,CH3-25);13C-NMR(100 MHz,C5D5N)數據見表1。以上數據與文獻[4,6]報道的3-O-β-D-吡喃木糖基-(1→3)-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃木糖基-齊墩果酸-28-O-β-D-吡喃葡萄糖苷比較,為該化合物經堿水解脫去28-葡萄糖基的次生皂苷,鑒定其為齊墩果酸-3-O-β-D-吡喃木糖基-(1→3)-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃木糖苷。
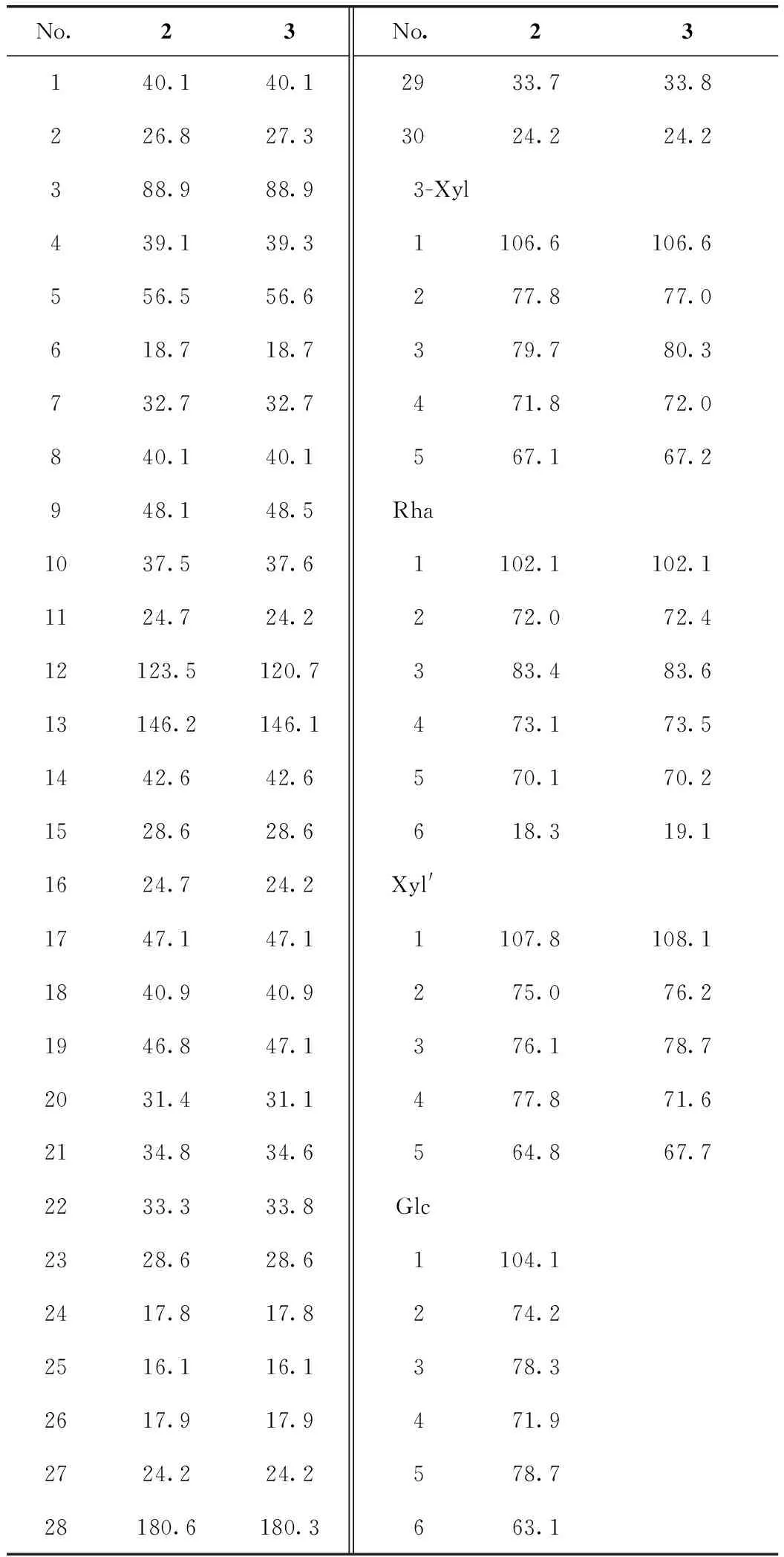
表1 化合物2和3的13C-NMR數據(100 MHz,C5D5N)
4 結論
敗醬中的化學成分復雜,對照品分離較為困難。本文首先采用大孔樹脂精制技術,能夠有效富集總皂苷;對其中的主要成分,先采用減壓硅膠柱色譜進行快速分段,再采用硅膠和反相硅膠柱色譜,分離純化各化合物;對難以分離的2個化合物PSH-2和PSH-3,采用反相高壓制備柱色譜技術進行分離;終端純化采用Sephadex凝膠柱色譜,進一步除去其他殘余雜質,提高產品純度。通過上述分離純化技術的綜合應用,最終成功獲得3種目標產物對照品。結果表明該分離方法較為簡潔、高效,為進一步開發利用黃花敗醬提供參考。
植物藥化學成分復雜,成分相對含量不一,因為質量研究中所需的特定對照品純度要求高,標定用量大,所以對其分離純化工作量大,獲取難度高。近年來,隨著多種分離填料以及制備用中壓和高壓液相色譜儀器的普及,分離工作帶得到了很大改善。本課題組研究表明,上述對照品制備技術作為一種通用流程,也可為其他高純度成分的制備提供借鑒[7-8]。
[1] 劉明生,陳英杰,卜月華,等.黃花敗醬研究進展[J].沈陽藥學院學報,1993,10(4):301-304.
[2] 高亮,張琳,劉江云,等.黃花敗醬的化學成分研究[J].中草藥,2011,42(8):1477-1480.
[3] Gao L,Zhang L,Li N,et al.New triterpenoid saponins fromPatriniascabiosaefolia[J].Carbohyd Res,2011,346(18):2881-2885.
[4] Gao L,Zhang L,Wang L M,et al.New triterpenoid saponins fromPatriniascabiosaefolia[J].J Asian Nat Prod Res,2012,14(4):333-341.
[5] Xu Q M,SHU Z,HE W J,et al.Antitumor activity ofPulsatillachinensis(Bunge)Regel saponins in human liver tumor 7402 cells in vitro and in vivo[J].Phytomedicine,2012,19(3-4):293-300.
[6] Tian J,Wu F E,Qiu M H,et al.Triterpenoid saponins fromPterocephalushookeri[J].Phytochemistry,1993,32(6):1535-1538.
[7] WANG L,Xu Q,Su S,et al.Simultaneous purification of Pulchinenoside B4and B5fromPulsatillachinensisusing macroporous resin and preparative high performance liquid chromatography[J].Ind Eng Chem Res,2012,51(45):14859-14866.
[8] 劉慧璐,馮建勇,王增尚,等.高色價梔子黃的精制工藝研究[J].中國現代應用藥學,2013,30(12):1315-1319.
SeparationofReferenceCompoundsfromTotalSaponinofPatriniascabiosaefolia
LUHao,LIUHongwei,WANGZengshang,LIUJiangyun*,HAOLili,YANGShilin
(CollegeofPharmaceuticalSciences,SoochowUniversity,Jiangsu,Suzhou,215123,China)
Objective:This study managed to develop an efficient separation process to afford 3 saponins as reference compounds fromPatriniascabiosaefoliaFisch.Methods:The process was conducted using multiple column chromatographies including silica gel,reversed-phase silica gel,sephadex gel columns,and preparative middle-/high-pressure liquid chromatography.The HPLC/ELSD method was applied for purity analysis of samples.Results:Three saponins1-3were obtained with their purities above 98.9%.Conclusion:The separation process developed was highly efficient and compact,which resulted in target compounds with high purities.
Patriniascabiosaefolia;saponin;reference compound;separation
10.13313/j.issn.1673-4890.2015.6.015
2014-11-17)
江蘇省科技支撐計劃社會發展項目(BE2012649)
*
劉江云,副教授,研究方向:天然藥物化學;Tel:(0512)65884301,E-mail:liujiangyun@suda.edu.cn
