脊髓性肌萎縮癥2個核心家系SMN1基因分析
2015-10-22 12:44:32曾光群楊季云張丁丁通訊作者
中國實用神經(jīng)疾病雜志 2015年23期
關(guān)鍵詞:檢測
曾光群 楊季云 張丁丁(通訊作者) 陳 蓉 李 寧
1)四川彭州市人民醫(yī)院 彭州 611930 2)四川省醫(yī)學(xué)科學(xué)院 四川省人民醫(yī)院人類疾病基因研究四川省重點實驗室 成都 610072 3)川北醫(yī)學(xué)院 南充 637000 4)遵義醫(yī)學(xué)院 遵義 563000
脊髓肌肉萎縮癥(spinal muscular atrophy,SMA)是僅次于囊性纖維化的兒童致死性疾病,是由于脊髓前角運動神經(jīng)元變性引起的漸進性近端肌肉無力和癱瘓的神經(jīng)肌肉性疾病,大多數(shù)為常染色體隱性遺傳,也有報道為常染色體顯性遺傳和X 連鎖遺傳[1]。發(fā)病率1/6 000~1/10 000,攜帶率在不同的人群中為1/40~1/60[2-3],我國南部正常人群此病的攜帶率約1/60[4],上海、臺灣正常人群此病的攜帶率約1/39[5]和1/48[6]。
1995年法國學(xué)者將SMA 的基因定位于5q11.2~13.3,認為反向重復(fù)的運動神經(jīng)元生存基因(survival motor neuron gene,SMN)為致病基因[7]。該基因有2個序列高度相似的拷貝:靠近端粒的決定性基因SMN1和靠近著絲粒的修飾基因SMN2,兩者間有5個堿基的區(qū)別,編碼序列內(nèi)僅1 個堿基差異。SMN 所在的染色體區(qū)域內(nèi)結(jié)構(gòu)復(fù)雜,且存在眾多重復(fù)序列和假基因簇,致其結(jié)構(gòu)不穩(wěn)定,發(fā)生缺失或轉(zhuǎn)換的頻率較高,使相應(yīng)的SMN1基因拷貝數(shù)復(fù)雜多變。95%以上的SMA 患者有SMN1第7號外顯子純合缺失,其余5%是SMN1點突變或者復(fù)合型的雜合缺失[1]。SMN2 基因的拷貝數(shù)是SMN1基因缺失的劑量補償,與患兒臨床表型的嚴重程度相關(guān)[8]。MLPA 是一種檢測基因缺失或重復(fù)的高效、準確方法[9],STR 連鎖能分析風(fēng)險染色體的來源。本研究聯(lián)合兩種方法對2個SMA 家系成員SMN1基因檢測,明確了基因攜帶情況,給患兒和家庭提供了完整的評估。
1 資料和方法
1.1 一般資料 先證者1,女,11歲,G1P1,足月順產(chǎn),圍生期無異常。3歲前運動無異常,自3歲開始出現(xiàn)下蹲困難,易摔等現(xiàn)象,下肢比上肢嚴重。體格檢查:身高130cm,四肢肌肉萎縮,肌力、肌張力及膝腱反射減弱,病理征未引出。頭顱MRI未見明顯異常,肌電圖表現(xiàn)為神經(jīng)源性損害。輔助檢查:磷酸肌酸激酶324U/L(參考值26.00~174.00U/L),磷酸肌酸激酶同工酶52U/L(參考值0~25.00U/L)。患兒父母體健,非近親結(jié)婚,家族中無同類病史。
先證者2,男4歲。G1P1,足月順產(chǎn),圍生期無異常。患兒自幼運動發(fā)育落后,2歲左右開始出現(xiàn)運動能力倒退,走路不穩(wěn),上下樓梯困難,下蹲后不易站起,呈鴨子步態(tài)。體格檢查:體型瘦小,雙側(cè)腓腸肌略肥大,四肢肌力Ⅳ級、肌張力降低,膝腱反射減弱,病理征陰性,Gower征(+)。輔助檢查:磷酸肌酸激酶408U/L,磷酸肌酸激酶同工酶62U/L,肌電圖呈典型的失神經(jīng)性改變。患兒父母體健,非近親結(jié)婚,家族中無同類病史,母親懷孕18 周。
1.2 研究方法
1.2.1 基因組DNA 提取:簽署知情同意書后,取患兒及家人乙二胺四乙酸鈉抗凝外周血2 mL,無菌抽取孕婦羊水5 mL。用DNA 抽提試劑盒(北京天根生化科技有限公司)提取基因組DNA,操作步驟嚴格按照試劑盒說明書進行。NanoDrop2000測定濃度。將DNA 濃度校正至50ng/μL,-20 ℃保存。
1.2.2 MLPA 檢測:按荷蘭MRC-Holland公司提供的MLPA 試劑盒檢測。DNA 樣本(50ng)5μL,經(jīng)變性、探針雜交過夜、連接和PCR 反應(yīng)等步驟后,取PCR 產(chǎn)物1.0μL,加1.5μL LIZ-500,HD 7μL,混勻,ABI3130xl測序儀進行片段分析[10]。
1.2.3 結(jié)果判定:使用GeneMapper軟件收集數(shù)據(jù),Coffalyser軟件進行分析。如無信號則為外顯子缺失,表現(xiàn)為相關(guān)峰消失,信號成倍增加表示發(fā)生重復(fù)。攜帶者相應(yīng)外顯子缺失信號降低35%~55%。
1.2.4 STR 基 因 連 鎖 分 析:根 據(jù) 文 獻[11],選 用4 個 連 鎖STR 位點(D5S435、D5S351、D5S610、D5S629)對2個家系分析。引物由上海生工合成,熒光標記引物兩翼,ABI3130xl遺傳分析儀上機檢測,進行單倍型連鎖分析。
2 結(jié)果
2.1 MLPA 結(jié)果 家系1中先證者SMN1基因第7號外顯子及8號外顯子拷貝數(shù)均為零,母親和外祖母SMN1基因第7號外顯子及8號外顯子為1個拷貝數(shù),其爺爺SMN1基因第7號外顯子及8號外顯子為3個拷貝數(shù)。父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數(shù)。家系2先證者SMN1基因第7號外顯子及8號外顯子拷貝數(shù)均為零,母親和胎兒SMN1基因第7號外顯子及8號外顯子為1個拷貝數(shù),父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數(shù)。
2.2 單倍型分析結(jié)果 家系1先證者與其父親有相同的母源致病的SMN1等位基因。家系2 先證者和胎兒均遺傳1條父親的致病的等位基因片段。
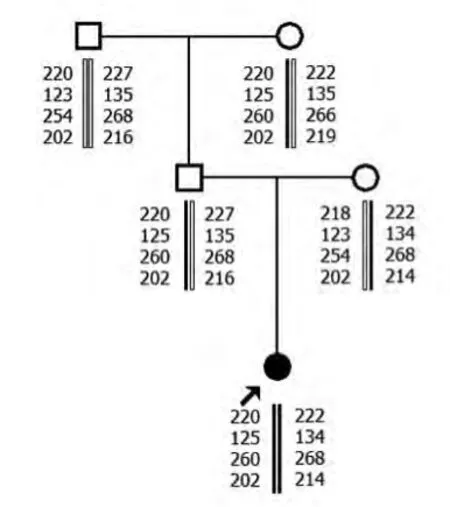
家系1
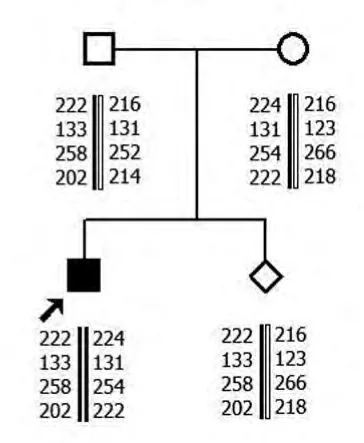
家系2
3 討論
SMA 的診斷主要依靠臨床表現(xiàn)、實驗室檢查、家族遺傳史及基因檢測4個方面,但臨床表現(xiàn)和常規(guī)輔助檢測缺乏特異性,故臨床上對確診SMA 更多依賴于基因檢測。臨床上SMA 各型間的差異很大,依據(jù)發(fā)病年齡和臨床病程嚴重程度的不同,分為Ⅰ型、Ⅱ型、Ⅲ型和Ⅳ型,其中Ⅰ型約占70%,臨 床 表 型 最 嚴 重,多 數(shù) 于6 個 月 內(nèi) 起 病,2 歲 內(nèi) 夭 折[12]。SMN1基因缺失(包括SMN1基因純合缺失或SMN1基因轉(zhuǎn)換為SMN2基因)或SMN1基因內(nèi)微小突變是引起SMA 的主要原因。SMN1基因缺失及其附近基因的大范圍缺失,引起的臨床表型較嚴重,SMN1基因轉(zhuǎn)換為SMN2基因,引起的臨床表型較輕,但SMN 基因的缺失和表型的嚴重程度缺失之間沒有相關(guān)性[13]。
每條染色體上有0~4個拷貝的SMN 基因,大部分純合缺失的患者SMN1拷貝數(shù)為0,健康人SMN1拷貝數(shù)為2,攜帶者和雜合缺失的患者SMNI拷貝數(shù)為1,約5%的正常個體可有3~4個SMN1拷貝,曲曉星等[5]報道在正常個體中有5~7個拷貝。因而通過檢測SMN1拷貝數(shù)可診斷SMA患者以及篩查攜帶者。SMA 攜帶者主要有4種基因型[14]:2條同源染色體上,1 條染色體上有1 拷貝正常的SMN1 基因,另1條缺失SMN1基因的為“1+0”型;1條染色體上有2拷貝SMN1基因,另1條缺失SMNI基因的“2+0”型;1條染色體上有1拷貝SMN1基因,另1條SMN1基因發(fā)生微小突變的“1+1m”型;1條染色體上有2拷貝SMNI基因,另1條SMN1基因發(fā)生突變的“2+1m”型;“1+0”型攜帶者最常見,約占95%,其他約占5%。
本研究先證者1和2SMN1基因的第7號外顯子及8號外顯子拷貝數(shù)均為零,可在基因水平診斷先證者為SMA。先證者1母親和外祖母SMN1基因第7號外顯子及8號外顯子為1個拷貝數(shù),先證者2母親SMN1基因第7號外顯子及第8號外顯子為1個拷貝數(shù),為雜合缺失突變,提示其均為攜帶者。先證者1爺爺SMN1 基因第7 號外顯子及第8號外顯子為3個拷貝數(shù),SMN1基因正常。先證者1和2父親SMN1基因第7號外顯子及8號外顯子為2個拷貝數(shù),表型上為正常。但其家中卻有患兒出現(xiàn),結(jié)合STR 分析,顯示先證者遺傳了1條父親致病的等位基因片段,為“2+0”SMA致病基因攜帶者,即1條染色體上有2拷貝SMN1基因,另1條染色體缺失SMN1基因。家系2胎兒SMN1基因第7號外顯子及8號外顯子為1個拷貝數(shù),1條為父源致病等位基因片段,1條為母源正常等位基因片段,為SMA 攜帶者,可以生育。
SMA 的嚴重程度由兩條染色體上攜帶突變的SMN 基因、SMN2基因拷貝數(shù)等其他因素決定。當前檢測SMA 攜帶者的方法,尚存在一定局限性。“1+1m”型和“2+0”型的SMA 攜帶者每條染色體上含SMN1相同劑量的拷貝數(shù),因此普通人群中檢測出含有2拷貝SMN1只能說明攜帶者的風(fēng)險降低,不能完全排除“1+1 m”型雜合缺失和“2+0”型SMA 攜帶者,在下一代中仍可有發(fā)病者。由于SMA 是一種發(fā)病率較高的遺傳性致死性疾病,基因攜帶率大,至今尚無有效的治療措施,產(chǎn)前診斷是預(yù)防該病的有效手段,因而患者的診斷以及攜帶者的檢出尤為重要。我們聯(lián)合MLPA 和STR 連鎖分析方法對2個核心家系SMN1基因分析,驗證了先證者的風(fēng)險等位基因的來源,檢出有2個拷貝的“2+0”型SMA 攜帶者,增加了檢出率。
通過以上分析,我們發(fā)現(xiàn)將MLPA 和STR 連鎖分析的聯(lián)合應(yīng)用,可充分發(fā)揮二者的優(yōu)勢,明確SMA 攜帶者,確診先證者。臨床上考慮為SMA 患者應(yīng)對以下情況進行SMN檢測:(1)有SMA 生育史;(2)自身為SMA 攜帶者,欲生育,則須對其配偶進行攜帶者檢測;(3)夫妻雙方家族均有脊髓性肌肉萎縮癥病史;(4)夫妻均為攜帶者或曾經(jīng)是脊髓性肌肉萎縮癥患者;(5)先證者經(jīng)基因診斷明確的高危產(chǎn)婦的。為保證產(chǎn)前診斷的準確性,應(yīng)結(jié)合多態(tài)性連鎖分析以減少風(fēng)險發(fā)生率。對于無SMN1 純合缺失的患者,對其基因內(nèi)微小突變檢測。對基因確診的患者,要分析5q13 區(qū)域內(nèi)其他修飾基因的變異,預(yù)測臨床表型和病情進展。
[1] Jiang W,Ji X,Xu Y,et al.Molecular prenatal diagnosis of autosomal recessive spinal muscular atrophies using quantification polymerase chain reaction[J].Genet Test Mol Biomarkers,2013,17(5):438-442.
[2] Markowitz JA,Singh P,Darras BT.Spinal muscular atrophy:a clinical and research update[J].Pediatr Neurol,2012,46(1):1-12.
[3] Kocheva SA,Plaseska-Karanfilska D,Trivodalieva S,et al.Prenatal diagnosis of spinal muscular atrophy in Macedonian families[J].Genet Test,2008,12(3):391-393.
[4] Prior TW.Spinal muscular atrophy diagnostics[J].J Child Neurol,2007,22(8):952-956.
[5] 曲曉星,肖冰,季星,等.應(yīng)用熒光定量PCR 開展上海地區(qū)脊肌萎縮癥攜帶者的人群篩查[J].中華醫(yī)學(xué)遺傳學(xué)雜志,2013,30(1):1-4.
[6] Su YN,Huang CC,Lin SY,et al.Carrier screening for spinal muscular atrophy(SMA)in107,611pregnant women during the Period 2005-2009:A prospective population-based cohort study[J].PloS One,2011,6(2):e17 067.
[7] Roy N,McLean MD,Besner-Johnston A,et al.Refined physical map of the spinal muscular atrophy gene(SMA)region at 5q13based on YAC and cosmid contiguous arrays[J].Genomics,1995,26(3):451-460.
[8] 盧麗萍,麻宏偉,姜俊,等.脊髓型肌萎縮臨床表型與基因拷貝數(shù)變化的相關(guān)性研究[J].中華遺傳學(xué)雜志,2007,24(2):144-147.
[9] Lalic T,Vossen RH,Coffa J,et al.Deletion and duplication screening in the DMD gene using MLPA[J].Eur J Hum Genet,2005,13(11):1 231-1 234.
[10] Schouten JP,McElgunn CJ,Waaijer R,et al.Relative quantification of 40nucleic acid sequences by multiplex ligation-dependent probe amplification[J].Nucleic Acids Res,2002,30(12):e57.
[11] 孫維,沈嘉瑋,龍飛,等.優(yōu)選短串聯(lián)重復(fù)序列應(yīng)用于脊髓肌萎縮癥產(chǎn)前診斷的連鎖分析[J].上海交通大學(xué)學(xué)報,2010,30(6):707-712.
[12] Zerres K,Rudnik-Schoneborn S.Natural history in proximal spinal muscular atrophy.Clinical analysis of 445patients and suggestions for a modification of existing classifications[J].Arch Neurol,1995,52(5):518-523.
[13] Dastur RS,Gaitonde PS,Khadilkar SV,et al.Correlation between deletion patterns of SMN and NAIP genes and the clinical features of spinal muscular atrophy in Indian patients[J].Neurol India,2006,54(3):255-259.
[14] Chen Wj,Wu ZY,Wang N,et al.Quantitative studies on SMN1gene and carrier testing of Spinal muscular atrophy[J].Chin J Med Genet,2005,22(6):599-602.
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
