重組內(nèi)肽酶AspN的原核表達(dá)純化和活性鑒定
2015-10-25 12:09:56鄭娟郭懷祖李晶段樹燕趙自葉張大鵬郭尚敬王皓
生物技術(shù)通報 2015年1期
鄭娟郭懷祖李晶段樹燕趙自葉張大鵬郭尚敬王皓
(1. 聊城大學(xué)藥學(xué)院,聊城 252000;2. 抗體藥物與靶向治療國家重點(diǎn)實驗室,上海 201203;3. 第二軍醫(yī)大學(xué)腫瘤研究所,上海 200433)
重組內(nèi)肽酶AspN的原核表達(dá)純化和活性鑒定
鄭娟1,2郭懷祖2,3李晶2,3段樹燕1,2趙自葉1,2張大鵬2,3郭尚敬1王皓2,3
(1. 聊城大學(xué)藥學(xué)院,聊城 252000;2. 抗體藥物與靶向治療國家重點(diǎn)實驗室,上海 201203;3. 第二軍醫(yī)大學(xué)腫瘤研究所,上海 200433)
蛋白內(nèi)肽酶AspN是一種鋅金屬內(nèi)切肽酶,能選擇性切割天冬氨酸的N端肽鍵,廣泛應(yīng)用于蛋白的多肽制備及質(zhì)量肽圖譜鑒定。目前內(nèi)肽酶AspN來源于細(xì)菌分泌,產(chǎn)量低,制備困難,成本高,極大地限制了該酶的應(yīng)用。將腦膜膿毒性黃桿菌分泌的蛋白內(nèi)肽酶AspN所對應(yīng)的基因克隆入表達(dá)載體pET32a,導(dǎo)入E.coli BL21(DE3),首次運(yùn)用原核表達(dá)系統(tǒng)進(jìn)行可溶性融合表達(dá),親和層析對重組蛋白進(jìn)行純化。用HPLC、SDS-PAGE和熒光底物Anthranilyl-Ala-Phe-Ala-Phe-Asp-Val-Phe(NO2)-Tyr-Asp對重組酶進(jìn)行酶活鑒定。結(jié)果表明重組的內(nèi)肽酶AspN具有與標(biāo)準(zhǔn)品基本一致的酶切活性,能夠較好地應(yīng)用于生物和制藥領(lǐng)域。
重組內(nèi)肽酶AspN;原核表達(dá);純化;活性鑒定
內(nèi)肽酶AspN(flavastacin EC=3.4.24.76)是一種鋅金屬內(nèi)切肽酶,能選擇性切割天冬氨酸殘基的N端肽鍵,其來源于腦膜膿毒性黃桿菌分泌表達(dá),含有443個氨基酸,分為3部分,即信號肽、前肽和具有酶切活性的flavastacin。其中第1-91位氨基酸是信號肽部分[1],通過這個信號肽腦膜膿毒性黃桿菌將該蛋白酶分泌出胞外[2,3];第190位谷氨酸是酶活性中心,第189位、193位和199位的組氨酸是鋅金屬結(jié)合位點(diǎn),起催化作用[4,5]。天然酶在革蘭氏陰性菌腦膜毒性黃桿菌分泌時含有O-連接的糖基化位點(diǎn)修飾[6],其具體功能尚不明確。該酶能夠選擇性切割天冬氨酸N端肽鍵[7],廣泛應(yīng)用于生物制藥和生產(chǎn)[8-10],特別是蛋白或多肽的制備以及蛋白的肽圖譜及質(zhì)量肽圖鑒定[11,12]。目前應(yīng)用的內(nèi)肽酶AspN主要來自于宿主菌的提取,其制備過程復(fù)雜,表達(dá)量低,純化困難,且純度不高,成本昂貴,極大地限制了該酶在生物及醫(yī)藥領(lǐng)域的應(yīng)用,近年來對該酶的研究尚未有突破的進(jìn)展。開發(fā)重組表達(dá)的工藝來進(jìn)行高效率低成本地制備蛋白內(nèi)肽酶AspN已經(jīng)成為解決當(dāng)前問題的關(guān)鍵[13-15]。大腸桿菌因其高效、廉價而成為重組蛋白的最佳宿主。大腸桿菌表達(dá)重組蛋白廣泛應(yīng)用于外源蛋白的表達(dá)[16-18]。然而大腸桿菌表達(dá)重組蛋白極易形成包涵體,即使是可溶性表達(dá)成活性酶,也會對宿主菌產(chǎn)生較大的毒性。故在本研究中通過Trx融合表達(dá)方式,既可以可溶性表達(dá),也可以在一定程度上抑制酶的活性,同時6個His標(biāo)簽利于蛋白的純化。本研究經(jīng)過全基因合成蛋白活性肽段所對應(yīng)的核苷酸序列,構(gòu)建AspN/pET32表達(dá)載體。首次采用Trx融合表達(dá)的方式進(jìn)行AspN的可溶性表達(dá),經(jīng)過鎳金屬螯合層析柱進(jìn)行一步親和層析獲得高表達(dá)的融合蛋白,然后通過緩沖液置換及酶的自激活從而制備出高純度的目的蛋白。運(yùn)用HPLC、SDS-PAGE和熒光試驗對重組酶進(jìn)行酶活鑒定,結(jié)果表明該重組AspN具有與和天然提取的標(biāo)準(zhǔn)品基本一致的酶切活性。這種重組內(nèi)肽酶AspN-Trx具有制備簡單,表達(dá)量、純度高的特點(diǎn),為該酶廣泛應(yīng)用于蛋白多肽制備、肽圖譜鑒定、晶體結(jié)構(gòu)研究及活性作用機(jī)制研究奠定了良好的基礎(chǔ)。
1 材料與方法
1.1 材料
1.1.1 載體和菌株 載體:原核表達(dá)載體pET/32a為實驗室保存;菌株:大腸桿菌TG1和BL21(DE3)均為實驗室傳代保存。
1.1.2 試劑與材料 限制性內(nèi)切酶BamH I和Xho I購自日本TaKaRa公司;T4 DNA連接酶購自美國Promega公司;DNA marker DL2000 Hind III為華美公司產(chǎn)品;蛋白marker購自Thermo Sciencific公司。小量質(zhì)粒抽提試劑盒、DNA膠回收試劑盒購自上海生工技術(shù)有限公司;Goldview購自北京天根生物技術(shù)有限公司;瓊脂糖購自上海實生細(xì)胞生物技術(shù)有限公司;細(xì)菌培養(yǎng)試劑:胰蛋白胨、酵母提取物、瓊脂粉購自英國Oxiod公司;氨芐青霉素粉末購自北京制藥有限公司;丙烯酰胺膠濃縮液、過硫酸銨、TEMED均購自上海博光生物技術(shù)有限公司;鎳金屬螯合層析柱和凝膠過濾層析G25 脫鹽柱均購自GE公司;熒光底物Anthranilyl-Ala-Phe-Ala-Phe-Asp-Val-Phe(NO2)-Tyr-Asp是通過上海紫域生物有限公司合成;標(biāo)準(zhǔn)內(nèi)肽酶AspN購自NEB公司;組蛋白為本實驗室自行提取,重組西妥昔單克隆抗體為本實驗室自行表達(dá)與純化。
1.2 方法
1.2.1 目的基因的獲取 文獻(xiàn)報道內(nèi)肽酶AspN是通過腦膜膿毒性黃桿菌分泌上清純化得到的[6]通過uniprot數(shù)據(jù)庫獲得目的蛋白的氨基酸序列。由于本研究運(yùn)用原核胞內(nèi)融合表達(dá)系統(tǒng),故將前端信號肽及前肽部分予以去除。成熟部分蛋白的氨基酸序列如下:

將氨基酸序列轉(zhuǎn)換為核苷酸序列并根據(jù)其在大腸桿菌翻譯中常用的密碼子進(jìn)行密碼子優(yōu)化,以避免稀有密碼子的出現(xiàn)。優(yōu)化后的核苷酸序列由Invitrogen公司合成。
1.2.2 表達(dá)載體的構(gòu)建 將全基因合成的基因片段從連接的pMD18-T載體上用限制性內(nèi)切酶BamHI和Xho I將目的片段酶切,膠回收目的片段;用T4 DNA連接酶將目的基因插入到BamH I和Xho I酶切的表達(dá)載體上,通過酶切鑒定和DNA測序篩選正確的克隆。
1.2.3 融合蛋白的誘導(dǎo)表達(dá) 將正確的重組質(zhì)粒轉(zhuǎn)化至表達(dá)菌株大腸桿菌BL21(DE3)。將含有重組質(zhì)粒的表達(dá)菌接種到試管擴(kuò)大培養(yǎng)至OD600為0.6左右時,菌液進(jìn)行分管誘導(dǎo),檢測誘導(dǎo)IPTG終濃度(0.01、0.05、0.1、0.2 mmol/L)、誘導(dǎo)溫度(16℃、25℃、30℃、37℃)、誘導(dǎo)時間(3、6、9 h 過夜)對重組內(nèi)肽酶AspN可溶性表達(dá)量的影響。每個時間點(diǎn),各取1 mL的菌液,離心棄上清,用200 μL 1× PBS懸浮,超聲破碎的菌離心后得到上清和沉淀。上清直接取樣,沉淀用200 μL 1×PBS懸浮。各取樣10 μL,非還原的樣品加入10 μL 2×loading buffer,還原樣品加入8 μL 2×loading buffer和2 μL 1 mol/L DTT。樣品95℃加熱5 min后,用12%的SDS-PAGE檢測蛋白表達(dá)情況。
1.2.4 融合蛋白的純化 按照最優(yōu)的誘導(dǎo)條件進(jìn)一步擴(kuò)大培養(yǎng)。接種含有200 μL AspN/pET32a重組質(zhì)粒的BL21菌接種至含Amp+的200 mL LB培養(yǎng)基中,37℃過夜培養(yǎng),次日以1∶20擴(kuò)大培養(yǎng)至OD600為0.6左右時,加入最優(yōu)濃度的IPTG,最優(yōu)的誘導(dǎo)溫度和時間。收集菌體,8 000 r/min,4℃離心,10 min,棄上清,得到的菌體沉淀。按質(zhì)量體積比1∶10用1×PBS進(jìn)行菌體懸浮。將混液置于冰上超聲破碎(功率360 W,超聲2 s,間歇5 s,共40 min)。然后11 000 r/min,4℃離心,30 min,分別收集上清和沉淀。上清用0.45 μm濾膜過濾后,通過鎳金屬螯合層析純化獲得帶有Trx標(biāo)簽的融合蛋白。平衡緩沖液為20 mmol/L PB,0.5 mol/L NaCl,20 mmol/L 咪唑(pH7.4);洗脫液為20 mmol/L PB,0.5 mol/L NaCl,0.5 mol/L 咪唑(pH7.4),采用階梯的洗脫方式進(jìn)行純化。目的融合蛋白由于Trx的存在,具有一定的酶活性抑制作用,在洗脫的緩沖液中,不會發(fā)生自切的現(xiàn)象。故最終得到的為重組內(nèi)肽酶AspN-Trx。該融合蛋白可經(jīng)凝膠過濾層析G25脫鹽柱將酶置換到最佳反應(yīng)緩沖液(50 mmol/L Tris,2.5 mmol/L ZnSO4,pH8.0)中發(fā)生自切,自動切除Trx融合標(biāo)簽而激活成完全活性的成熟酶。
1.2.5 HPLC檢測重組內(nèi)肽酶AspN的活性 空白組為重組內(nèi)肽酶AspN-Trx與組蛋白按1∶100混合后加甲酸立即終止反應(yīng)的樣品;試驗組為重組內(nèi)肽酶AspN-Trx與組蛋白按1∶100混合加入緩沖液(50 mmol/L Tris,2.5 mmol/L ZnSO4,pH8.0),37℃反應(yīng)過夜后樣品。將樣品離心后,分別取10 μL,用C18柱進(jìn)行HPLC檢測。
1.2.6 SDS-PAGE 檢測重組內(nèi)肽酶AspN的活性空白組為10 μL重組西妥昔單克隆抗體和激活的酶,對照組重組西妥昔單克隆抗體與酶混合后甲酸立即終止反應(yīng)。試驗組重組酶和重組西妥昔單克隆抗體按1∶10加入到緩沖液(50 mmol/L Tris,2.5 mmol/L ZnSO4,pH8.0),37℃反應(yīng)過夜。分別取各樣品10 μL,加入到含有8 μL 2×loading buffer 2 μL 1 mol/L DTT的上樣緩沖液中,95℃加熱5 min后,將20 μL樣品全部上樣,進(jìn)行12%SDS-PAGE檢測。
1.2.7 熒光試驗檢測重組內(nèi)肽酶AspN的活性 取1 mg熒光底物Anthranilyl-Ala-Phe-Ala-Phe-Asp-Val-Phe(NO2)-Tyr-Asp加入到1 mL的超純水中,完全溶解。取1 μL(約1 μmol)的熒光底物加入到含有不同酶量的內(nèi)肽酶AspN的150 μL緩沖液(50 mmol/L Tris,2.5 mmol/L ZnSO4,pH8.0)中,37℃反應(yīng),用330 nm的激發(fā)光,450 nm的發(fā)射光,熒光酶標(biāo)儀檢測熒光量。
2 結(jié)果
2.1 目的基因的獲取
由Invitrogen公司合成,共1 086個核甘酸序列如下:


2.2 表達(dá)載體的構(gòu)建
構(gòu)建的重組表達(dá)載體用BamH I和Xho I做雙酶切鑒定,獲得大小5 900 bp的線性pET32a表達(dá)載體和大小約1 000 bp的目的基因條帶(圖1)。結(jié)果表明成功構(gòu)建重組質(zhì)粒AspN/pET32a。
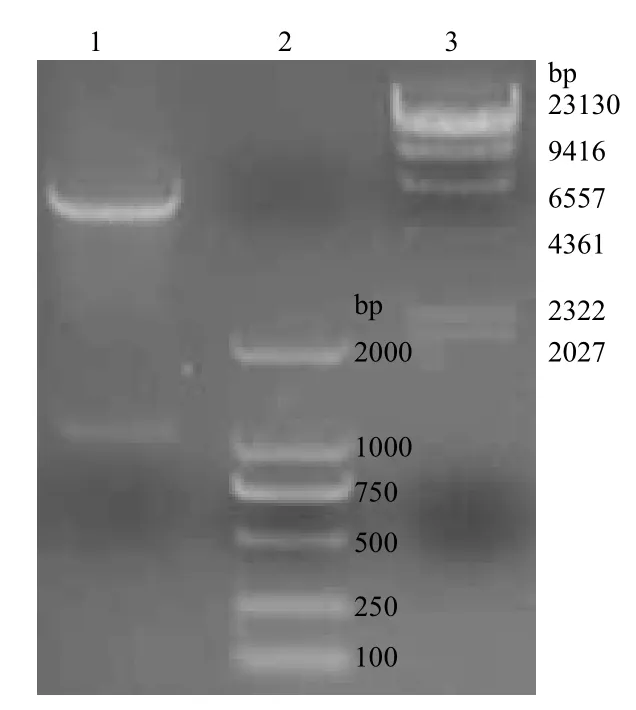
圖1 重組質(zhì)粒AspN/pET32a 的BamHI與XhoI雙酶切鑒定
2.3 融合蛋白的誘導(dǎo)表達(dá)
構(gòu)建正確的質(zhì)粒AspN/pET32a轉(zhuǎn)化至大腸桿菌BL21(DE3)表達(dá)菌株,通過IPTG誘導(dǎo)表達(dá)出與預(yù)期目的蛋白大小一致的重組蛋白,大小為57 kD。此融合蛋白以可溶性及包涵體的形式同時存在。由于包涵體復(fù)性過程復(fù)雜,并且復(fù)性得率不高,故繼續(xù)研究可溶性表達(dá)。為了避免重組酶發(fā)生自切,選擇低溫16℃;IPTG濃度影響不大,選擇常規(guī)0.1 mmol/L;避免誘導(dǎo)時間過長蛋白發(fā)生降解,選擇誘導(dǎo)6 h。最終確定在0.1 mmol/L IPTG,16℃,6 h的條件下獲得可溶的目的蛋白(圖2)。
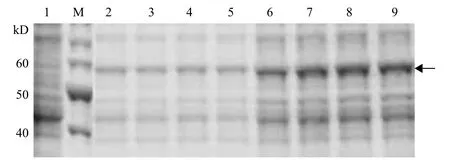
圖2 不同IPTG濃度AspN/pET32a的表達(dá)分析
2.4 融合蛋白的純化
SDS-PAGE檢測結(jié)果(圖3)顯示,在30%洗脫液(20 mmol/L PB,0.5 mol/L NaCl,150 mmol/L咪唑)的條件下得到目的蛋白。
經(jīng)誘導(dǎo)表達(dá)后的4.8 L的菌液經(jīng)親和層析和脫鹽的后得到4.5 mg,純度為90%左右的成熟內(nèi)肽酶AspN(圖4)。重組內(nèi)肽酶AspN純化過程中的得率和純化步驟,見表1。
2.5 HPLC檢測重組內(nèi)肽酶AspN的活性
通過重組內(nèi)肽酶AspN酶切組蛋白鑒定其活性。空白組重組酶AspN-Trx與組蛋白混合后立即終止反應(yīng),組蛋白特定的峰存在,而試驗組AspN-Trx與組蛋白37℃反應(yīng)過夜后組蛋白內(nèi)部的天冬氨酸N端的肽鍵被切斷,特定的組蛋白片段被切碎消失,如箭頭所標(biāo)示(圖5),說明重組內(nèi)肽酶AspN-Trx經(jīng)緩沖液自激活后具有較好的酶切活性。
2.6 SDS-PAG檢測重組內(nèi)肽酶AspN的活性
通過重組內(nèi)肽酶AspN切抗體鑒定其活性。重組酶與重組西妥昔單克隆抗體反應(yīng),檢測抗體特異的片段消失(圖6)說明重組內(nèi)肽酶AspN有較好的酶切活性。
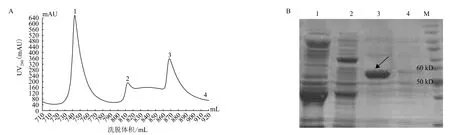
圖3 重組內(nèi)肽酶AspN-Trx鎳金屬螯合層析柱純化圖(A)和SDS-PAGE分析(B)
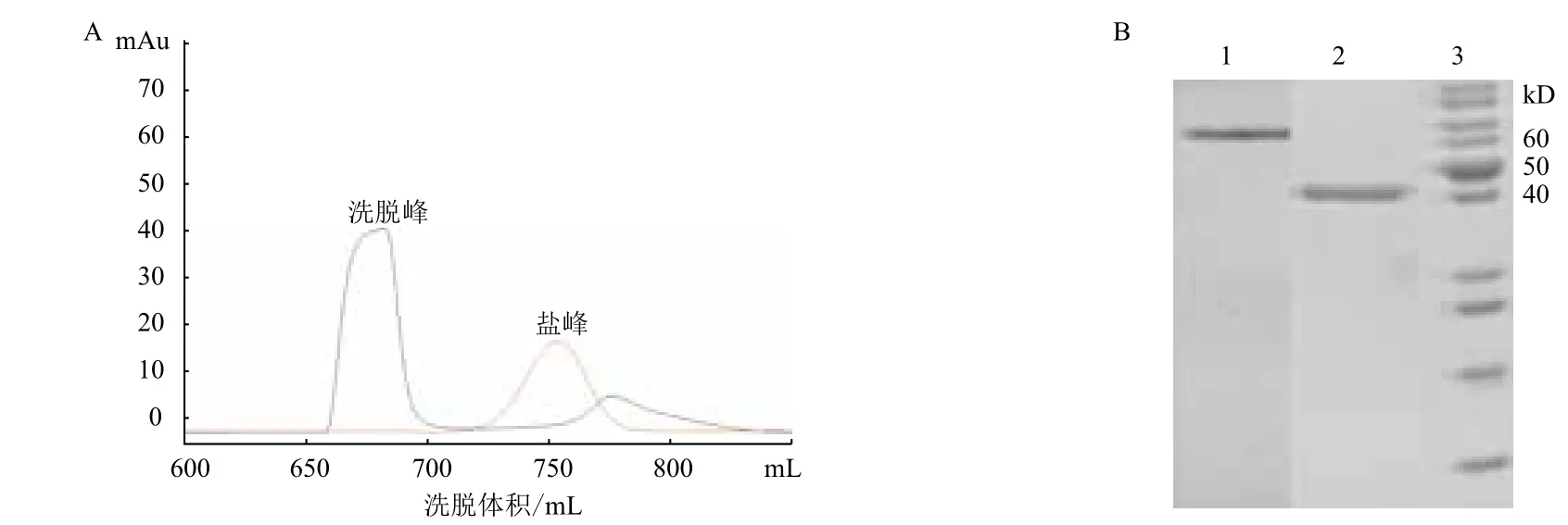
圖4 重組內(nèi)肽酶AspN-Trx凝膠過濾層析切除標(biāo)簽純化圖(A)和SDS-PAGE分析(B)

表1 重組內(nèi)肽酶AspN純化過程的得率和純化
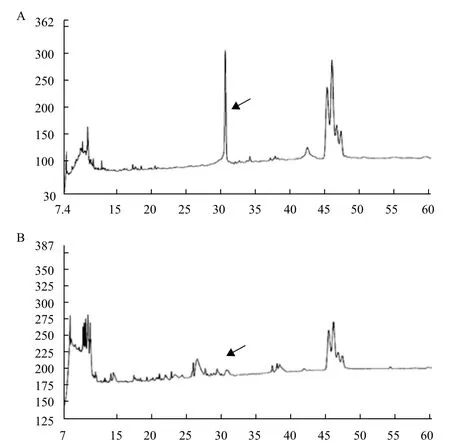
圖5 HPLC驗證重組內(nèi)肽酶AspN-Trx酶切活性
2.7 熒光試驗檢測重組內(nèi)肽酶AspN的活性
用熒光底物檢測蛋白的活性,底物濃度為飽和狀態(tài),通過反應(yīng)速率來衡量酶的活性(圖7)。重組內(nèi)肽酶AspN-Trx與標(biāo)準(zhǔn)品內(nèi)肽酶AspN具有基本一致的酶切活性。
3 討論
隨著內(nèi)肽酶AspN在生物和醫(yī)藥領(lǐng)域的廣泛應(yīng)用,通過宿主菌進(jìn)行表達(dá)提取的方式越來越成為瓶頸,開發(fā)重組表達(dá)工藝進(jìn)行高效率低成本的制備越發(fā)顯得必要。在本研究首次原核可溶性表達(dá)重組內(nèi)肽酶AspN,簡單的純化步驟即獲得了純度高活性好的內(nèi)肽酶AspN,其為進(jìn)一步進(jìn)行該酶晶體結(jié)構(gòu)研究和活性作用機(jī)制研究奠定了基礎(chǔ)。
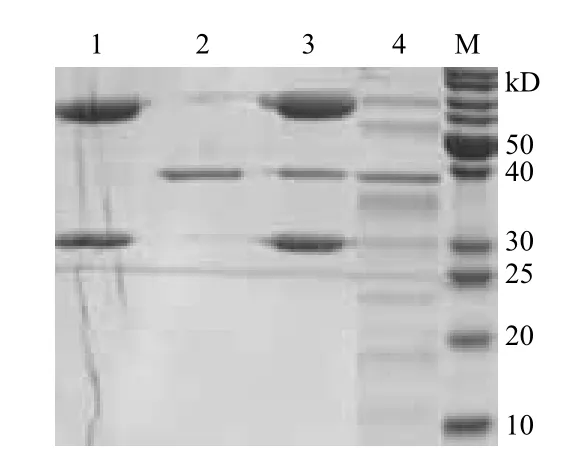
圖6 重組內(nèi)肽酶Asp酶活檢測的SDS-PAGE圖
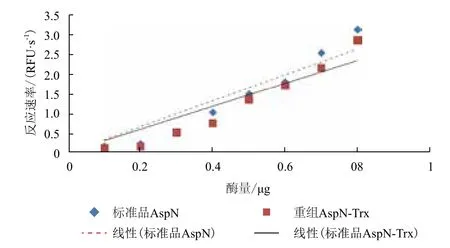
圖7 重組內(nèi)肽酶AspN-Trx與標(biāo)準(zhǔn)內(nèi)肽酶AspN的熒光試驗測活曲線
具有酶切活性的重組AspN通過Trx融合表達(dá)的方式來進(jìn)行可溶性表達(dá),其Trx既可以作為純化標(biāo)簽及易于蛋白空間結(jié)構(gòu)形成進(jìn)行可溶性高效表達(dá),同時也可以在一定程度上對該酶的活性進(jìn)行抑制,從而減少可溶性表達(dá)酶活高,對宿主菌造成毒性反應(yīng)。同時本研究也在誘導(dǎo)蛋白表達(dá)、收集菌體、超聲破碎菌體、離心獲得可溶表達(dá)的上清的過程中采用低溫條件,提高蛋白的穩(wěn)定性,并有效抑制酶的活性。在誘導(dǎo)過程中,IPTG濃度對總蛋白的表達(dá)量有一定影響,從0.01 mmol/L到0.1 mmol/L總蛋白表達(dá)條帶依次增高,0.1 mmol/L到0.2 mmol/L時增加不明顯,雖然從蛋白表達(dá)條帶上看,增加的主要是不溶性包涵體,最終選擇采用了0.1 mmol/L的誘導(dǎo)條件;由于該酶采用融合表達(dá)的方式,其融合標(biāo)簽抑制了酶的活性,所以在融合蛋白表達(dá)過程中優(yōu)先考慮適宜菌生長的37℃,有利于菌的繁殖獲得更多的目的蛋白,然而對于其它類型的活性酶來說要避開酶的最適溫度以免發(fā)生自切或是對宿主菌造成危害;在誘導(dǎo)表達(dá)的時間的選擇上,一般不宜過長,時間長菌的代謝物積累,一些宿主菌內(nèi)源性酶也會導(dǎo)致一些目的蛋白的降解,不利于目的蛋白的表達(dá)。在純化過程中,可采用分步洗脫多種方式,從而有效的得到較高濃度和純度的目的蛋白。
重組內(nèi)肽酶AspN-Trx在酶切緩沖液中發(fā)揮自激活后具有較高的酶切活性,經(jīng)過熒光底物活性檢測,該酶具有和對照提取酶基本一致的活性,由于天然酶在革蘭氏陰性菌腦膜毒性黃桿菌分泌表達(dá)中含有O-連接的糖基化位點(diǎn)修飾,大腸桿菌重組表達(dá)時缺少這種翻譯后修飾,而它們的活性差別不是太大,也就說明該O-糖基化修飾可能對酶的活性影響不大。若將該重組酶和天然酶進(jìn)行晶體結(jié)構(gòu)的解析將進(jìn)一步探索該糖基化修飾對該酶的空間結(jié)構(gòu)影響以及酶活中心和與底物的作用機(jī)制。
大腸桿菌表達(dá)外源蛋白具有產(chǎn)量高的優(yōu)點(diǎn),加上純化標(biāo)簽可以快速得到目的蛋白,同時利用HPLC、SDS-PAGE和熒光試驗檢測蛋白的活性具有快捷方便的特點(diǎn)。利用基因工程的手段,合理的試驗技術(shù)能夠快速、有效的得到有活性的蛋白酶,該酶的重組表達(dá)也為其它活性酶的重組表達(dá)提供了一個很好的范本。
4 結(jié)論
本研究首次利用基因工程技術(shù)在大腸桿菌里重組可溶性融合表達(dá)內(nèi)肽酶AspN,該重組酶AspN/ pET32a具有高表達(dá)的同時,還可以通過親和層析對該酶進(jìn)行快速地純化。運(yùn)用熒光試驗對酶進(jìn)行定量分析,重組內(nèi)肽酶具有與提取內(nèi)肽酶基本一致的酶切活性。
[1] Tarentino AL, Quinones G, Grimwood BG, et al. Molecular cloning and sequence analysis of flavastacin:an O-glycosylated prokaryotic zinc metalloendopeptidase[J]. Arch Biochem Biophys, 1995, 319(1):281-285.
[2] Tarentino AL, Quinones G, Trumble A, et al. Molecular cloning and amino acid sequence of peptide-N4-(N-acetyl-beta-D-glucosaminyl)asparagine amidase from flavobacterium meningosepticum[J]. J Biol Chem, 1990, 265(12):6961-6966.
[3] Tarentino AL, Quinones G, Schrader WP, et al. Multiple endoglycosidase(Endo)F activities expressed by Flavobacterium meningosepticum. Endo F1:molecular cloning, primary sequence,and structural relationship to Endo H[J]. J Biol Chem, 1992, 267(6):3868-3872.
[4] Tarentino AL, Quinones G, Changchien LM, et al. Multiple endoglycosidase F activities expressed by Flavobacterium meningosepticum endoglycosidases F2 and F3. Molecular cloning,primary sequence, and enzyme expression[J]. J Biol Chem, 1993,268(13):9702-9708.
[5] Tarentino AL, Quinones G, Hauer CR, et al. Molecular cloning and sequence analysis of Flavobacterium meningosepticum glycosylasparaginase:a single gene encodes the alpha and beta subunits[J]. Arch Biochem Biophys, 1995, 316(1):399-406.
[6] Reinhold BB, Hauer CR, Plummer TH, et al. Detailed structural analysis of a novel, specific O-linked glycan from the prokaryote Flavobacterium meningosepticum[J]. J Biol Chem, 1995, 270(22):13197-13203.
[7] Ingrosso D, Fowler AV, Bleibaum J, et al. Specificity of endoproteinase Asp-N(Pseudomonas fragi):cleavage at glutamyl residues in two proteins[J]. Biochem Biophys Res Commun, 1989, 162(3):1528-1534.
[8] Carulli S, Calvano CD, Palmisano F, et al. MALDI-TOF MS characterization of glycation products of whey proteins in a glucose/ galactose model system and lactose-free milk[J]. J Agric Food Chem, 2011, 59(5):1793-1803.
[9] Meltretter J, Pischetsrieder M. Application of mass spectrometry for the detection of glycation and oxidation products in milk proteins[J]. Ann N Y Acad Sci, 2008, 1126:134-140.
[10] Meltretter J, Seeber S, Humeny A, et al. Site-specific formation of Maillard, oxidation, and condensation products from whey proteins during reaction with lactose[J]. J Agric Food Chem, 2007, 55(15):6096-6103.
[11] Seidler J, Adal M, Kubler D, et al. Analysis of autophosphorylation sites in the recombinant catalytic subunit alpha of cAMP-dependent kinase by nano-UPLC-ESI-MS/MS[J]. Anal Bioanal Chem,2009, 395(6):1713-1720.
[12] Neubert H, Gale J, Muirhead D. Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS:assay development for total salivary pepsin/pepsinogen[J]. Clin Chem,2010, 56(9):1413-1423.
[13] Cohen SN, Chang AC, Boyer HW, et al. Construction of biologically functional bacterial plasmids in vitro[J]. Proc Natl Acad Sci USA,1973, 70(11):3240-3244.
[14] Itakura K, Hirose T, Crea R, et al. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin[J]. Science, 1977, 198(4321):1056-1063.
[15] Morrow JF, Cohen SN, Chang AC, et al. Replication and transcription of eukaryotic DNA in Escherichia coli[J]. Proc Natl Acad Sci U S A, 1974, 71(5):1743-1747.
[16] Sinsuwan S, Yongsawatdigul J, Chumseng S, et al. Efficient expression and purification of recombinant glutaminase from Bacillus licheniformis(GlsA)in Escherichia coli[J]. Protein Expr Purif, 2012, 83(1):52-58.
[17] Sarduy ES, Munoz AC, Trejo SA, et al. High-level expression of Falcipain-2 in Escherichia coli by codon optimization and autoinduction[J]. Protein Expr Purif, 2012, 83(1):59-69.
[18] Retnoningrum DS, Pramesti HT, Santika PY, et al. Codon optimization for high level expression of human bone morphogenetic protein-2 in Escherichia coli[J]. Protein Expr Purif, 2012, 84(2):188-194.
(責(zé)任編輯 李楠)
Cloning,Expression,Purification and Characterization of Soluble Recombinant Endoproteainase AspN
Zheng Juan1,2Guo Huaizu2,3Li Jing2,3Duan Shuyan1,2Zhao Ziye1,2Zhang Dapeng2,3Guo Shangjing1Wang Hao2,3
(1. School of Pharmacy of Liaocheng University,Liaocheng 252000;2. State Key Laboratory of Antibody Medicine and Targeted Therapy,Shanghai 201203;3. Second Military Medical University,Shanghai 200433)
Endoproteinase AspN(flavastacin)is a zinc metalloendopeptidase which can selectively cleave peptide bonds N-terminal to aspartic acid residues, also it is one of most widely used proteolytic enzyme used for protein digestion prior to MS or HPLC analysis. The natural endoproteinase AspN was extract from the bacteria, Elizabethkingia meningoseptica. Some factors including low yields, difficulty in purification and high cost, greatly limit the application of the enzyme. Target gene was cloned into an pET32a expression vector and transformed into E.coli BL21(DE3). This is the first report for the soluble expression of AspN-Trx in prokaryotic expression system and a poly-histidine tag enabled purification by Ni affinity chromatography. The activity of the recombinant endopeptidase AspN was identified by HPLC, SDS-PAGE and the fluorogenic substrate Anthranilyl-Ala-Phe-Ala-Phe-Asp-Val-Phe(NO2)-Tyr-Asp. The results showed that the recombinant endopeptidase AspN was consistent with the standard enzyme and can be used widely.
recombinant endoproteinase AspN;prokaryotic expression;purification;activity assay
10.13560/j.cnki.biotech.bull.1985.2015.01.028
2014-07-04
鄭娟,女,研究生,研究方向:分子細(xì)胞生物學(xué);E-mail:zhengjuan2006400741@126.com
郭尚敬,男,教授,碩士生導(dǎo)師,研究方向:基因工程藥物;E-mail:guoshangjing@lcu.edu.cn王皓,男,研究員,博士生導(dǎo)師,研究方向:基因工程抗體;E-mail:hwang_summu@163.com
猜你喜歡
中學(xué)生數(shù)理化·中考版(2022年8期)2022-06-14 06:55:24
今日農(nóng)業(yè)(2021年19期)2022-01-12 06:16:36
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年11期)2021-08-22 03:15:44
中學(xué)生數(shù)理化(高中版.高考數(shù)學(xué))(2021年1期)2021-03-19 08:28:38
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
無線電工程(2020年11期)2020-10-29 01:25:46
