鹽酸頭孢吡肟的合成
2015-10-28 05:45:27彭東明王曉紅劉艷飛尹偉成劉珍寶張航張姍姍
中南大學學報(自然科學版) 2015年7期
彭東明,王曉紅,劉艷飛,尹偉成,劉珍寶,張航,張姍姍
?
鹽酸頭孢吡肟的合成
彭東明1, 2,王曉紅1,劉艷飛1,尹偉成2,劉珍寶3,張航1,張姍姍1
(1. 中南大學化學化工學院,湖南長沙,410083;2. 湖南中醫藥大學藥學院,湖南長沙,410208;3. 中南大學藥學院,湖南長沙,410013)
以7-氨基頭孢烷酸(7-ACA)為原料,二氯甲烷和環己烷為溶劑,三甲基氯硅烷(TMSCl)為保護劑,與N-甲基吡咯烷(NMP)和三甲基碘硅烷(TMSI)制成的季銨鹽中間體反應,脫保護后成鹽得中間體7-氨基-3-(1-甲基-1-四氫吡咯)甲基-3-頭孢-4-羧酸鹽酸鹽(7-MPCA),再與苯并噻唑硫醇活性酯反應,合成目標產物鹽酸頭孢吡肟。研究硅烷化反應、碘代反應和成鹽反應的主要影響因素,進行工藝參數優化。采用紅外光譜、元素分析、核磁共振譜和質譜對產物鹽酸頭孢吡肟進行結構表征。研究結果表明:該工藝原材料易得,反應條件溫和,且操作簡單,以7-ACA的質量計鹽酸頭孢吡肟的總收率為74.6%。
7-氨基頭孢烷酸;鹽酸頭孢吡肟;藥物合成;抗生素
鹽酸頭孢吡肟(cefepime),商品名為馬斯平(maxipime),化學名為氯化1-[[(6R,7R)-7-[(2Z)-(2-氨基-4-噻唑基)-2-(甲氧亞氨基)乙酰氨基]-2-羧基-8-氧代-5-硫雜-1-氮雜雙環[4,2,0]-辛-2-烯-3-基]甲基]-1-甲基吡咯烷鎓一鹽酸鹽一水合物。1993年在瑞典首次上市,鹽酸頭孢吡肟作為第4代氨噻肟型頭孢菌素類抗生素,與第3代頭孢菌素相比具有更廣的抗菌譜[1?2],對革蘭氏陽性菌、陰性菌和厭氧菌均有良好的抗菌活性,并且對鏈球菌、肺炎鏈球菌的抗菌活性有所增強,能夠較好地治療急性呼吸道感染[3?5],人們對其進行了廣泛研究[6?8]。已報道的鹽酸頭孢吡肟的合成路線較多,如Aburaki等[9?10]以7-ACA為原料,得到的產物為兩性離子,此兩性離子在室溫下不穩定,且最后一步縮合的產率較低,中間體的分離必須過柱,操作繁瑣,進行工業化生產成本過高。Walker等[11]以7-ACA為原料, 氟利昂(Freon TF)為溶劑, 經雙三甲基硅化物中間體“一勺燴”得頭孢吡肟關鍵中間體7-氨基-3-(1-甲基-1-四氫吡咯)甲基-3-頭孢-4-羧酸鹽酸鹽(7-MPCA),收率為38%(以7-ACA計),化合物7-MPCA再與苯并噻唑硫醇活性酯進行7位酰化反應制得頭孢吡肟的鹽酸鹽,總收率為30.8%。該合成路線條件溫和,反應選擇性較好,但反應產率不高,使用氟利昂不符合環保要求。Lim等[12?14]對鹽酸頭孢吡肟的合成工藝進行研究,均以7-ACA為原料,經“一勺燴”合成中間體7-MPCA再與活性酯進行反應制得頭孢吡肟的鹽酸鹽,其中宮平等[13]所得總收率為24.7%,安明等[14]所得總收率為59.4%。本文作者在文獻[11]的基礎上,參考有關文獻[12?18],對其合成工藝進行了優化和改進:1) 以7-ACA為原料,用三甲基氯硅烷(TMSCl)代替傳統的六甲基二硅胺烷(HMDS)作為7-ACA的羧基和氨基的保護劑;2) 用二氯甲烷和環己烷代替氟利昂為反應溶劑;3) 將N-甲基吡咯烷(NMP)和三甲基碘硅烷(TMSI)制成季銨鹽中間體再與保護好的7-ACA反應制得關鍵中間體7-MPCA。
1 實驗部分
1.1 試劑
實驗所用試劑為:7-氨基頭孢烷酸(河北中潤制藥有限公司);三甲基氯硅烷(CP,武漢寶龍化工有限公司);N-甲基吡咯烷(AR,浙江臺州清泉醫藥化工有限公司);環己烷(AR,天津市大茂化學試劑廠);二氯甲烷(AR,湖南師大化學制劑廠);三乙胺(AR,天津市泰興試劑廠);甲醇(AR,天津市泰興試劑廠);苯并噻唑硫醇活性酯(AR,國產工業品);丙酮(AR,長沙市湘科精細化工廠);薄層層析硅膠(CP,青島海洋化工有限公司)。
1.2 合成路線
以7-ACA為原料改進后的合成鹽酸頭孢吡肟的反應路線如圖1所示。
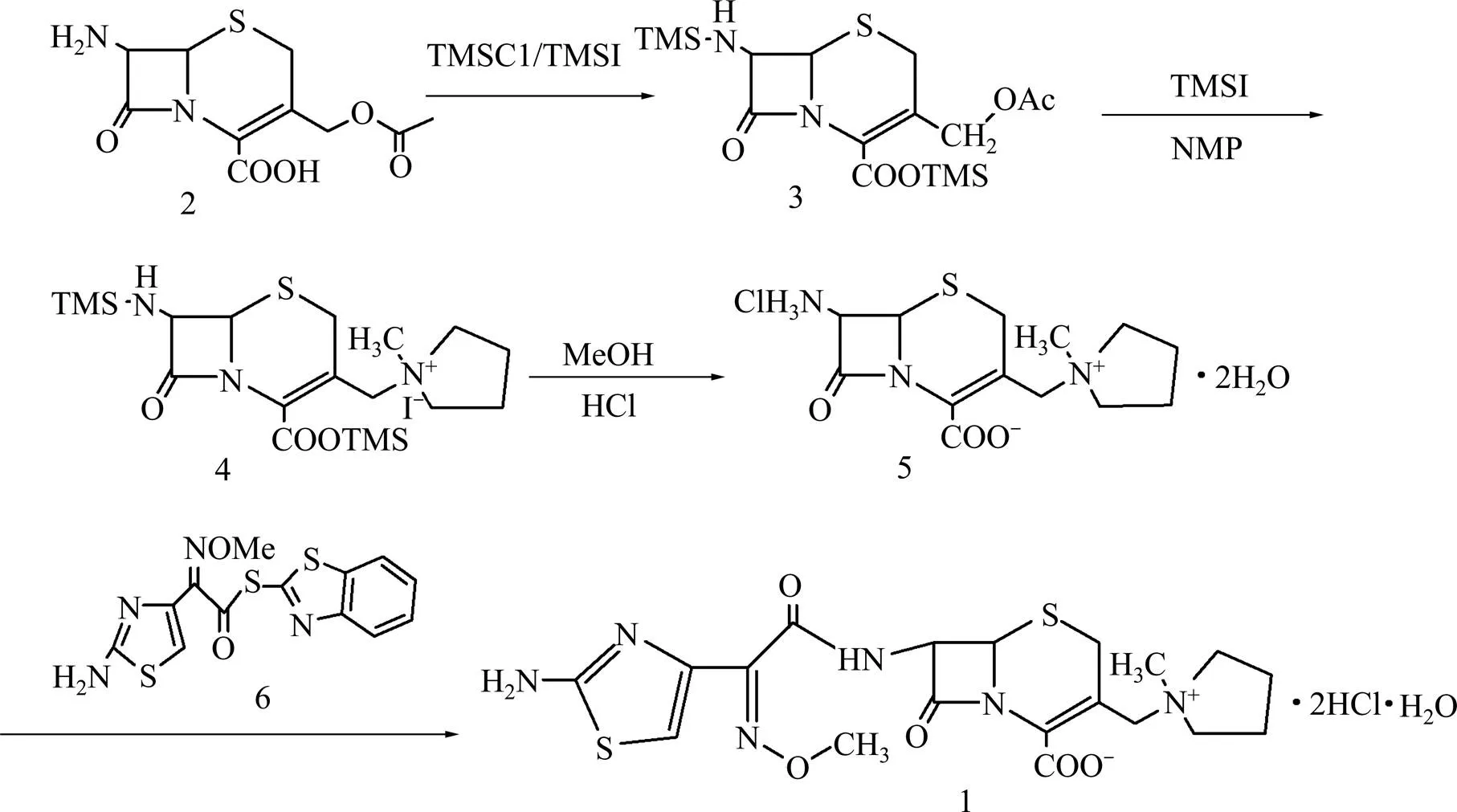
圖1 鹽酸頭孢吡肟的合成路線
1.3 操作步驟
1.3.1 化合物3的合成
往裝有回流冷凝管、干燥管和溫度計的150 mL三口燒瓶中加入7-ACA 5.0 g (18.4 mmol)、無水溶劑二氯甲烷或環己烷40 mL,于35 ℃下通入氮氣5 min,再加入TMSCl 5.41 g (49.7 mmol)和TMSI 0.11 g (0.6 mmol),在氮氣保護的條件下保持該溫度反應,采用薄層色譜(TLC)監測反應完全后,減壓蒸餾蒸出溶劑,得化合物3(見圖1,下同)。
化合物3的1H-NMR(DMSO,500 MHz)化學位移:0.23(s,9H,N-Si(CH3)3),0.38(s,9H,COOSi(CH3)3),1.51(d,1H,=13.6 Hz,NH),2.09(s,3H,COCH3),3.41(q,2H,=18.3 Hz,SCH2),3.61(q,2H,=18.3 Hz,SCH2),4.80(dd,1H,=4.5,13.6 Hz,C-7-lactam),4.83(q,2H,=13.2 Hz,CH2OAc),5.11(q,2H,=13.2 Hz,CH2OAc),4.91(d,1H,=4.5 Hz,C-6-lactam)。
1.3.2 化合物5的合成
往干燥的三口燒瓶中加入10 mL二氯甲烷,緩慢加入NMP 2.92 g (29.4 mmol)和TMSI 5.52 g (27.6 mmol),于25 ℃下攪拌2 h,在氮氣保護下冰水浴冷卻至5 ℃以下,緩慢加入化合物3,不斷攪拌并控制反應體系溫度低于10 ℃,隨后加入5 mL N,N-二乙基苯胺在5 ℃下繼續反應2 h,得化合物4。反應結束后,緩慢滴加2.5 mL無水甲醇,控制滴加速度使反應體系溫度低于10 ℃,滴加完畢后于5 ℃攪拌10 min,加入濃度為3.21 mol/L的鹽酸20 mL,加完后快速升溫至25 ℃,快速攪拌至完全分層;靜置后,分出水相,往水相中加入適量的活性炭于5 ℃攪拌下脫色30 min,過濾。量取濾液體積并轉入250 mL三口燒瓶中,快速攪拌下滴加5倍濾液體積的丙酮,在冰箱中放置過夜,析晶后過濾,并收集析出的固體;用預先冷卻的混合液丙酮與水的體積比即(丙酮):(水)=5:1洗滌,得淡黃色粉末,真空干燥后稱量,得5.80 g淡黃色晶體,收率為85.3%,HPLC測得其純度為95.3%。
化合物5的1H-NMR(500MHz,D2O)化學位移δ:2.14~2.32(m,4H,+N(CH3)CH2CH2CH2CH2),3.00 (s,3H,+NCH3),3.46~3.67(m,5H,+N(CH3)CH2CH2CH2- CH2,SCH2),3.96(q,1H,=16.9 Hz,SCH2),4.09(q,2H,=13.9 Hz,—CH2-+N(CH3)CH2CH2CH2CH2)、4.73(q,2H,=13.9 Hz,—CH2-+N(CH3)CH2CH2-CH2- CH2),5.21(d,1H,=5.1 Hz,C-7-lactam),5.41(d,1H,=5.1 Hz,C-6-lactam)。
1.3.3 鹽酸頭孢吡肟(1)的合成
在250 mL的三口燒瓶中加入5.80 g (17.3 mmol)化合物5和116 mL混合溶劑,在氮氣保護下攪拌,保持溫度在5 ℃以下,滴加三乙胺1.93 g (19.0 mmol);待固體全部溶解,控制pH在6.5左右,分次加入苯并噻唑硫醇活性酯6.10 g (12.5 mmol),于5 ℃下攪拌,TLC跟蹤至反應完全。
將反應液轉移至分液漏斗中,用蒸餾水萃取后合并水相并加入適量活性炭脫色30 min。將水相過濾并將濾液轉入體積為2 L的三口瓶中冰水浴冷卻至0~5 ℃,在攪拌下滴加鹽酸調節pH至1.0~2.0,量取濾液體積,于5 ℃下加入10倍濾液體積的丙酮并攪拌1 h,冷卻過夜,過濾并用混合溶液(丙酮):(水)=5:1洗滌,得微黃色固體粉末,真空干燥后稱質量,得7.82 g淡黃色晶體,收率為87.4%(以7-ACA為原料計總收率為74.6%),HPLC測其純度為99.3%。
鹽酸頭孢吡肟(1):1H-NMR(500MHz,D2O)化學位移:2.07~2.14(m,4H,+N(CH3)CH2CH2CH2CH2),2.95(s,3H,+NCH3),3.43~3.66(m,5H,+N(CH3)- CH2CH2CH2CH2,SCH2),3.93(s,3H,OCH3),4.02(q,1H,=17.5Hz,SCH2),4.32(q,2H,=13.5 Hz,—CH2-+N(CH3)CH2CH2CH2CH2),4.61(q,2H,=13.5 Hz,—CH2-+N(CH3)CH2CH2CH2CH2),5.33(d,1H,=5.0 Hz,C-7-lactam),5.87(d,1H,=5.0 Hz,C-6-lactam),6.87(s,1H,SCH),7.40~9.96(s,2H,NH2),9.78(d,1H,=8.0 Hz,NH)
2 結果與討論
2.1 硅烷化反應
2.1.1 硅烷化試劑的選擇
在合成頭孢菌素類化合物時,通常要對7-ACA,ACLE及其類似物的4位羧基或7位氨基的活潑氫進行保護。常見保護羧基的方法是將其制成酯類,常用的保護氨基的方法是將其轉化成胺的硅衍生物,但需在無水條件下制備。由于7-ACA需同時對氨基和羧基進行保護,故一般選擇硅烷化試劑。傳統的保護劑為六甲基二硅胺烷(HMDS),雖可有效保護氨基和羧基,但其活性較低,一般要在60~80 ℃下回流8~13 h,反應溫度較高,反應時間較長,容易發生副反應。本實驗采用價格較低且活潑性較強的三甲基氯硅烷代替傳統的六甲基硅烷對7-ACA氨基和羧基同時進行保護,在降低生產成本的同時,大大縮短了反應時間,有利于實現大規模工業化生產。
2.1.2 硅烷化試劑物質的量對反應的影響
在回流反應中保護劑TMSCl會伴隨溶劑的揮發逸出反應體系,為保證反應完全,TMSCl的物質的量要超過理論值。為確定TMSCl與7-ACA的物質的量之比,在其他條件不變的情況下,實驗僅改變TMSCl的物質的量,觀察反應3 h后7-ACA轉化率的情況,結果如表1所示。
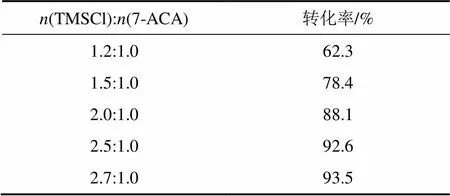
表1 n(TMSCl):n(7-ACA)對7-ACA轉化率的影響
由表1可知:在其他條件相同時,不同物質的量的TMSCl對該步反應影響較大,當(TMSCl):(7-ACA)=2.7:1時,其轉化率最高,可達93.0%以上。
2.1.3 硅烷化溶劑對反應的影響
選用環己烷和二氯甲烷作溶劑來代替對環境有破壞作用的Freon TF,在節約成本的同時有效降低了對環境的破壞。在其他條件相同的情況下,加入5.0 g 7-ACA,考察不同溶劑對7-ACA轉化的影響,結果如表2所示。

表2 不同溶劑對7-ACA轉化率的影響
由表2可以看出:該步反應采用二氯甲烷或環己烷為溶劑與采用Freon TF為溶劑的轉化率相近,但更經濟、環保。
2.2 碘代反應
為提高產品收率,實驗中將TMSI與NMP先制成季銨鹽再與化合物3進行反應。因此,TMSI與NMP的物質的量之比為影響該步反應的關鍵因素,在其他條件不變的情況下,用不同物質的量比下制得的季銨鹽與化合物3反應,結果如表3所示。

表3 n(TMSI):n(NMP)對化合物3轉化率的影響
由表3可知:在其他條件相同時,隨NMP物質的量的增加,化合物3的轉化率也增加,當(TMSI):(NMP)=1:1.1時,該步反應轉化率達90.2%,繼續增加NMP的物質的量,化合物3的轉化率并沒有顯著提高。所以,當(TMSCl):(NMP)=1.0:1.1時,其轉化率較高,可達90.0%以上。
2.3 水解和成鹽反應
反應中一般需要對氨基和羧基進行保護,一般采用醇解和酸解來解除保護。先用甲醇對反應物進行水解,再選擇一定濃度的鹽酸進行水解,得中間體7-MPCA的鹽酸鹽。而中間體7-MPCA結構中的β-內酰胺環在酸堿環境下都易開環,因此,中間體的鹽酸水溶液飽和程度對其有較大影響。若鹽酸的濃度太小,則不能使其成鹽的pH滿足要求;若鹽酸的濃度太大,則會破壞中間體的結構。因此,鹽酸的濃度對其成鹽有較大影響。研究發現:當鹽酸濃度為1.07 mol/L和2.14 mol/L時,加入鹽酸攪拌后均有固體殘留,中間體不能完全成鹽酸鹽;當鹽酸的濃度為3.21 mol/L時,加入鹽酸攪拌后固體完全溶解,中間體可以完全形成鹽酸鹽。
2.4 鹽酸頭孢吡肟的合成
從中間體7-MPCA為原料,采用“一鍋法”合成化合物1。該步反應是酯的氨解,為親核?消除反應。為進一步考查溶劑對反應的影響,在相同條件下,分別采用不同溶劑進行反應。通過實驗可得:當采用(水):(丙酮)=1:2混合溶劑時,鹽酸頭孢吡肟的收率為78.4%;采用二氯甲烷為溶劑時,其收率為72.2%;采用(二氯甲烷):(甲醇)=52:9為溶劑時,其收率最高為88.2%。
2.5 結構確證
2.5.1 元素分析
采用元素分析儀對鹽酸頭孢吡肟(1)樣品元素的質量分數進行分析,結果如表4所示。
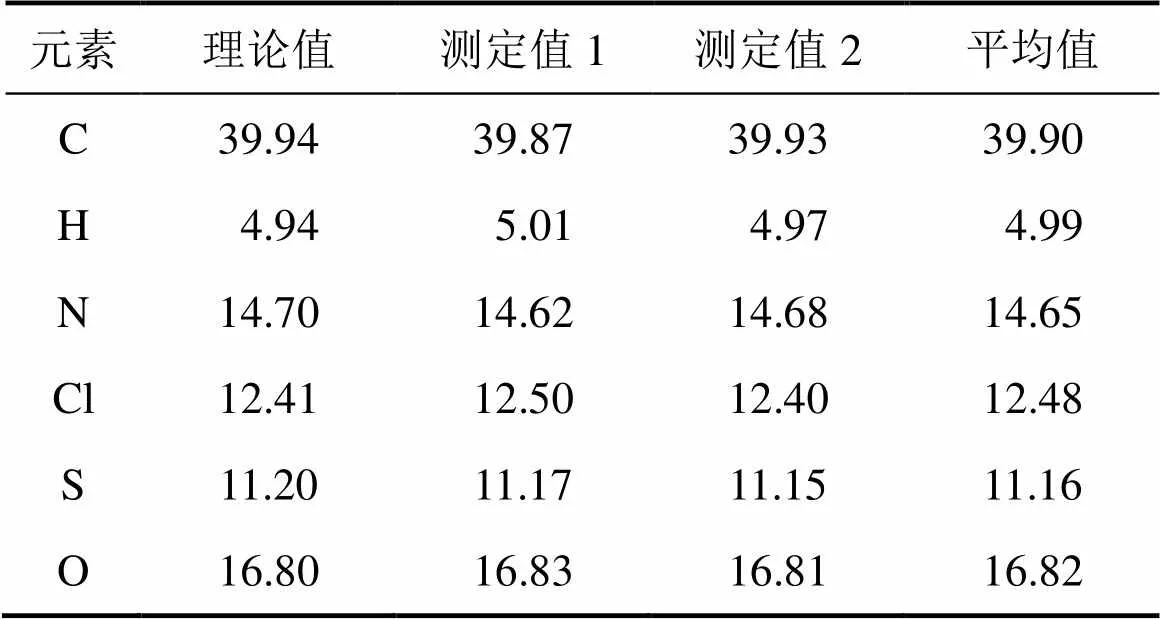
表4 鹽酸頭孢吡肟的元素分析結果(質量分數)
由表4可知:各元素的質量分數的實測值與理論值基本一致;沉淀滴定法的分析結果證實Cl以陰離子的形式存在于結構中,故合成樣品的化學式可表述為C19H24N6O5S2·2HCl·H2O,與鹽酸頭孢吡肟的化學式一致。
2.5.2 紫外光譜(UV)
合成的鹽酸頭孢吡肟(1)的紫外光譜分析結果如表5所示。

表5 鹽酸頭孢吡肟的紫外光譜分析結果
注:中性溶液指質量濃度為0.027 85 mg/L的甲醇溶液;酸性溶液指含0.1 mol HCl的質量濃度為0.026 66 mg/L 的甲醇溶液;堿性溶液指含0.1 mol NaOH的質量濃度為0.023 56 mg/L 的甲醇溶液。
由表5可知:鹽酸頭孢吡肟(1)中有明顯的K吸收帶,且在堿性溶液中K吸收帶顯著紫移,這與鹽酸頭孢吡肟結構中和官能團的共軛吸收特征相符。
2.5.3 質譜(MS)
合成鹽酸頭孢吡肟(1)樣品的質譜圖如圖2所示。鹽酸頭孢吡肟的化學式為C19H24N6O5S2·2HCl·H2O,相對分子質量為571.33。由圖2可見:在質譜中,沒有出現HCl和H2O的峰,因此,不計HCl和H2O的相對分子質量可得鹽酸頭孢吡肟堿基的質荷比為480。圖中質荷比481為[M+H]的分子離子峰,396為[M+H]的分子離子峰失去基團()后形成的碎片離子的質荷比。
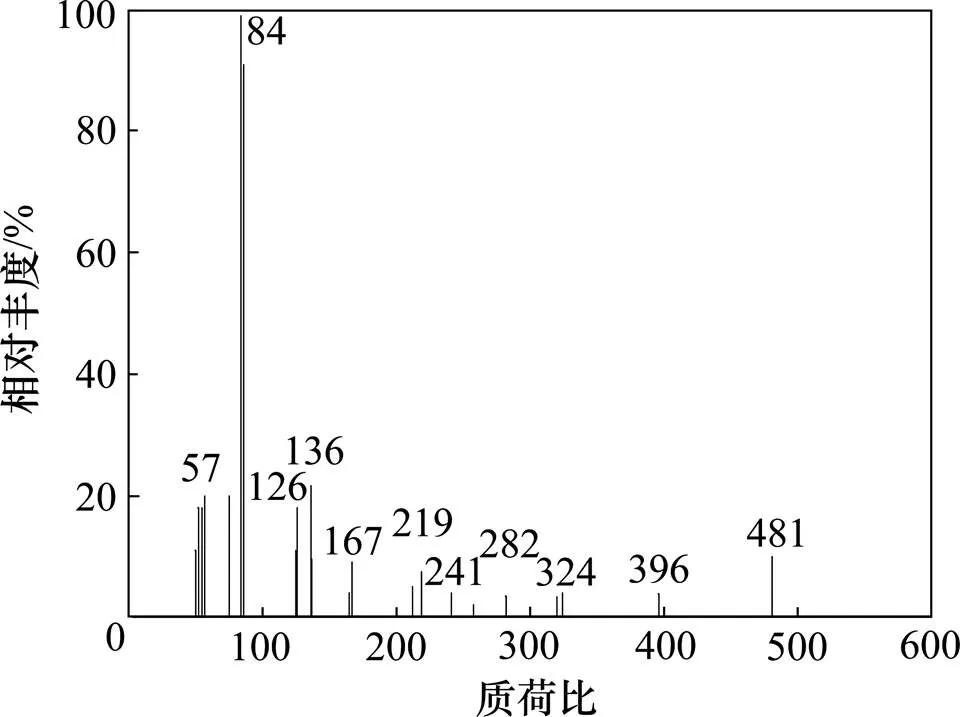
圖2 化合物1的MS譜
質荷比為324,282,241,219和84各碎片峰都可找到相對應的碎片離子,其中質荷比為84的碎片峰因相對豐度最大為基峰。鹽酸頭孢吡肟(1)的質譜分析結果如表6所示。

表6 化合物1 MS分析結果
2.5.4核磁共振譜(1H-NMR)
鹽酸頭孢吡肟(1)的核磁共振氫譜如圖3所示。
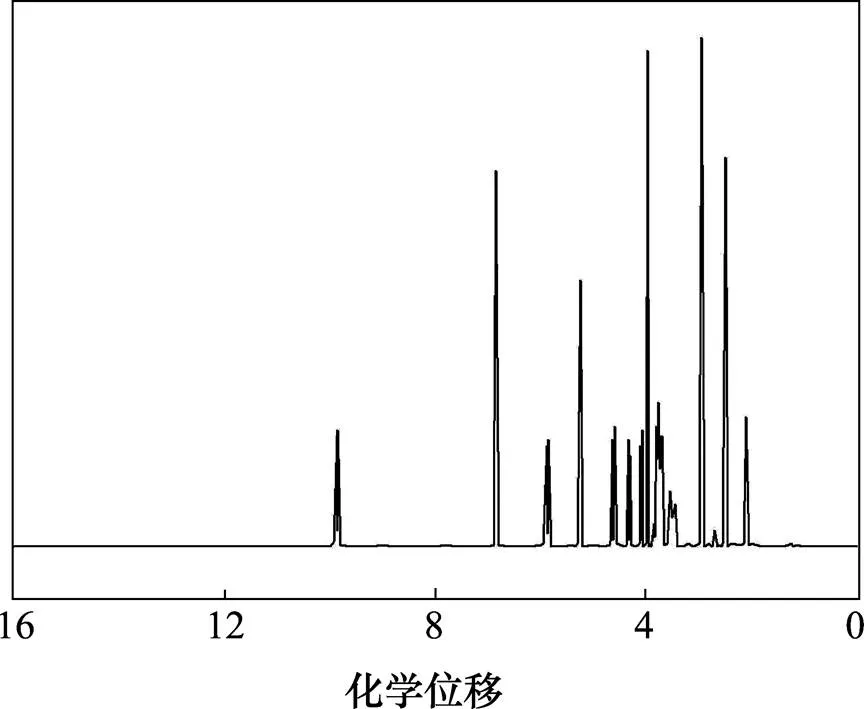
圖3 化合物1的1H-NMR譜圖
由圖3可知:1H-HMR譜中有13組峰,峰面積積分比為1:1:1:1:1:1:1:1:3:1:4:3:4,除去H(NH2),H(2HCl)和H(H2O)形成極寬峰不易辨認外,其他各峰的化學位移、質子個數和耦合常數等都與鹽酸頭孢吡肟的結構特征相符。按結構式(如圖4所示)不同位置的氫譜分析結果如表7所示。
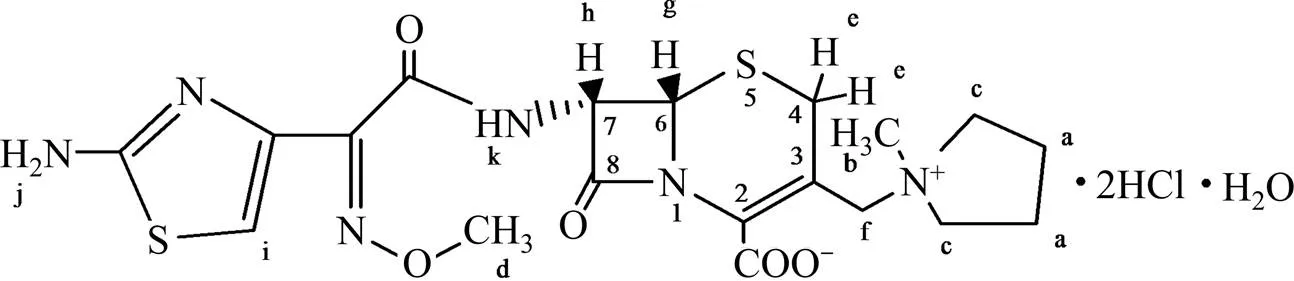
圖4 化合物1的結構圖
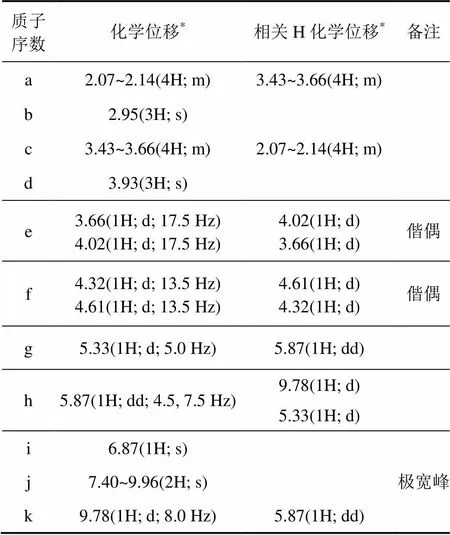
表7 鹽酸頭孢吡肟的1H-NMR譜測定結果
*:括號中含質子個數、峰的信息(s指單峰;d指雙重峰;m指多重峰;dd指雙二重峰)及耦合常數。
3 結論
1) 本實驗以7-ACA為原料,二氯甲烷和環己烷為溶劑,三甲基氯硅烷為保護劑,與N-甲基吡咯烷和三甲基碘硅烷制成季銨鹽反應,脫保護后成鹽合成鹽酸頭孢吡肟的關鍵中間體7-MPCA,再與苯并噻唑硫醇活性酯反應,最終生成鹽酸頭孢吡肟,該合成路線操作簡單,對環境污染小,原料易得,產品純度高,產品總收率能達到74.6%。
2) 以三甲基氯硅烷代替傳統的HMDS作硅烷化試劑,縮短了反應時間,減少了副反應的發生,有效提高了工藝效率。
3) 將N-甲基吡咯烷和三甲基碘硅烷制成季銨鹽,克服了N原子電子云密度高、反應活性強、副反應多的弊端,提高了產品的收率和純度。
4) 通過工藝優化得到了最佳的反應條件如下:(TMSCl):(7-ACA)=2.7:1.0,(TMSI):(NMP)=1.0:1.1;硅烷化反應溫度為35 ℃,反應時間3 h;由7-MPCA合成鹽酸頭孢吡肟,采用的溶劑為二氯甲烷與甲醇混合液的體積比即(二氯甲烷):(甲醇)=52:9。
參考文獻:
[1] Ohki H, Kawabata K, Inamoto Y, et al. Studies on 3′-quaternary ammonium cephalospoins-Ⅲ synthesis and antibacterial activity of 3′-(3-aminopyrazolium) cephalosporins[J]. Bioorganic & Medicinal Chemistry, 1997, 5(3): 557?567.
[2] 薛雨, 陳宇瑛. 頭孢菌素類抗生素的最新研究進展[J]. 中國抗生素雜志, 2011, 36(2): 86?92. XUE Yu, CHEN Yuying. New development of cephalosporin antibiotics[J]. Chinese Journal of Antibiotics, 2011, 36(2): 86?92.
[3] 龔軼欣. 鹽酸頭孢吡肟治療急性呼吸道感染的效果觀察[J]. 中外醫療, 2013, 32(9): 108?110. GONG Yixin. The effect of cefepime hydrochloride in treatment of acute respiratory tract infection[J]. China & Foreign Medical Treatment, 2013, 32(9): 108?110.
[4] 趙賈漪. 鹽酸頭孢吡肟治療小兒下呼吸道感染的療效觀察[J]. 醫學信息, 2014(5): 310. ZHAO Jiayi. The effect of cefepime hydrochloride in treatment of acute the children with lower respiratory infection[J]. Medical Information, 2014(5): 310.
[5] Behin S, Punitha I S R, Krishnan S. Stability studies of cefepime hydrochloride by stability indicating RP-HPLC method[J]. International Journal of Pharmaceutical Sciences and Nanotechnology, 2013, 6(3): 2181?2186.
[6] LI Yu, XU Li, WANG Fuan, et al. Solubilities of cefepime hydrochloride in water (Ethanol, 1-Propanol, or 2-Propanol) from (278.15 to 308.15) K [J]. Journal of Chemical & Engineering Data, 2010, 55(9): 4098?4103.
[7] 彭東明, 王春燕, 劉艷飛, 等. 鹽酸頭孢吡肟緩釋微球的制備與工藝優化[J]. 中南大學學報(自然科學版), 2013, 44(3): 907?913. PENG Dongming, WANG Chunyan, LIU Yanfei, et al. Preparation and optimization of controlled release microspheres of cefepime dihydrochloride[J]. Journal of Central South University (Science and Technology), 2013, 44(3): 907?913.
[8] 郝軍香, 李謙和, 彭東明. 頭孢吡肟的合成進展[J]. 合成化學, 2004, 12(1): 43?48. HAQ Junxiang, LI Qianhe, PENG Dongming. Development in synthesis of cefepime[J]. Chinese Journal of Synthetic Chemistry, 2004, 12(1): 43?48.
[9] Aburaki S, Kamachi H, Narita Y, et al. Cephalosporin: DE 3307550[P]. 1983?09?27.
[10] Aburaki S, Kamachi H, Narita Y, et al. Cephalosporins: US 4525473[P]. 1983?06?25.
[11] Walker D G, Brodfuehrer P R, et al. Use of bistrimethylisilylated intermediates in the preparation of semisynthetic 7-amino-3- substituted-cephems. Expedient synthesis of a new 3-[(1-methyl- 1-pyrrolidinio) methyl] cephalosporin[J]. Journal of Organic Chemistry, 1988, 53(5): 983?991.
[12] Lim G M F, Rouble J M. Process for the preparation of a cephalosporin antibiotic: US 15594129[P]. 1997?01?14.
[13] 宮平, 趙燕芳, 馮潤良, 等. 鹽酸頭孢吡肟的合成[J]. 中國藥物化學雜志, 2002, 12(6): 350?352. GONG Ping, ZHAO Yanfang, FENG Runliang, et al. Synthesis of cefepime dihydrochloride[J]. Chinese Journal of Medicinal Chemistry, 2002, 12(6): 350?352.
[14] 安明, 常珍, 高雷, 等. 鹽酸頭孢吡肟的合成[J]. 中國醫藥工業雜志, 2004, 35(9): 515?516. AN Ming, CHANG Zhen, GAO Lei, et al. Synthesis of cefepime hydrochloride [J]. Chinese Journal of Pharmaceuticals, 2004, 35(9): 515?516.
[15] 周磊, 樊會芳. 一種鹽酸頭孢吡肟的合成方法: CN 101735251 A[P]. 2009?12?26. ZHOU Lei, FAN Huifang. A synthesis method of cefepime hydrochloride: CN 101735251 A[P]. 2009?12?26.
[16] Page N, Stevenson R, Powell M. Analysis of N-methylpyrrolidine in cefepime hydrochloride by ion chromatography using suppressed conductivity detection with solid-phase extraction pre-treatment[J]. The Royal Society of Chemistry, 2014, 6(4): 1248?1253.
[17] 唐雙強. 第四代頭孢菌素鹽酸頭孢吡肟及中間體合成條件的研究[D]. 石家莊: 河北科技大學化學與制藥工程學院, 2008: 14?18. TANG Shuangqiang. Study on the synthetical technnics of cefepime hydrochloride and it’s intermediate[D]. Shijiazhuang: Hebei University of Science and Technology. College of Chemistry and Pharmaceutical Engineering, 2008: 14?18.
[18] Subramanian N H, Thyagarajan S, Manigandan P, et al. An improved ion chromatography method for fast and sensitive determination of N-methylpyrrolidine in cefepime hydrochloride[J]. Journal of Chromatography Science, 2009, 47(8): 549?552.
Synthesis of cefepime hydrochloride
PENG Dongming1, 2, WANG Xiaohong1, LIU Yanfei1, YIN Weicheng2, LIU Zhenbao3, ZHANG Hang1, ZHANG Shanshan1
(1. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China; 2. School of Pharmacy, Hunan University of Chinese Medicine, Changsha 410208, China;3. School of Pharmacy, Central South University, Changsha 410083, China)
The intermediate 7-MPCA was obtained by being salified after deprotection based on the reaction of the intermediate of quaternary ammonium salt produced by N-methyl pyrrolidine (NMP) and trimethyliodosilane (TMSI), and 7-aminocephalosporanic acid (7-ACA) was used as raw material, dichloromethane and cyclohexane as solvents, and trimethylchlorosilane (TMSCl) as the protective agent. Cefepime hydrochloride was synthesized from 7-amino-3-[(1-methyl-1-pyrrolidino) methyl]-3-cephem-4-carboxylic acid (7-MPCA) with benzothiazolethiol active ester. The main influencing factors of reactions such as silylation, iodization and salification were analyzed, and the process parameters were optimized. The structure of cefepime hydrochloride was confirmed by IR, elemental analysis, NMR and MS. The results show that the improved synthetic method has some advantages, such as easy availability of raw materials, mild reaction condition, and simple operation. The total yield of cefepime hydrochloride is 74.6% based on the mass fraction of 7-ACA.
7-aminocephalosporanic acid; cefepime hydrochloride; pharmaceutical synthesis; antibiotics
10.11817/j.issn.1672-7207.2015.07.003
R914.5
A
1672?7207(2015)07?2405?07
2014?11?24;
2015?01?13
國家自然科學基金資助項目(81301258);湖南省科技計劃項目(2012SK3134);湖南省自然科學基金資助項目(13JJ3099) (Project(81301258) supported by the National Natural Science Foundation of China; Project(2012SK3134) supported by Science and Technology Program of Hunan Province; Project(13JJ3099) supported by the Natural Science Foundation of Hunan Province)
劉艷飛,博士,副教授,從事藥物合成研究;E-mail: liuyf@csu.edu.cn
(編輯 劉錦偉)
