快速溶劑萃取-超高效液相色譜-串聯質譜法測定蜂花粉中氯霉素
2015-11-05 05:45:33祝子銅雷美康陳玉嬌章應俊衢州出入境檢驗檢疫局綜合技術服務中心浙江衢州324002
食品工業科技 2015年20期
祝子銅,雷美康,彭 芳,李 東,陳玉嬌,章應俊(衢州出入境檢驗檢疫局綜合技術服務中心,浙江衢州324002)
快速溶劑萃取-超高效液相色譜-串聯質譜法測定蜂花粉中氯霉素
祝子銅,雷美康,彭芳,李東,陳玉嬌,章應俊
(衢州出入境檢驗檢疫局綜合技術服務中心,浙江衢州324002)
建立了快速溶劑萃取-超高效液相色譜-串聯質譜法(UPLC-MS/MS)測定蜂花粉中氯霉素的分析方法。樣品以乙酸乙酯為萃取溶劑經快速溶劑萃取儀(ASE)萃取,硅膠固相萃取柱凈化,采用液質聯用多反應監測負離子模式測定,同位素內標法定量。方法線性范圍0.5~10 ng/mL,相關系數為R2=0.9936,回收率在88.3%~110.0%之間,相對標準偏差在5.3%~8.9%之間,檢出限為0.1 μg/kg。該方法具有效率高、試劑用量少、選擇性好、靈敏度高等優點。
蜂花粉,快速溶劑萃取,超高效液相色譜-串聯質譜法,氯霉素
氯霉素是一種應用廣泛的廣譜抗生素,對各類家禽、家畜、水產品及蜜蜂的各種傳染性疾病的控制和治療起重要作用[1]。同時,氯霉素具有嚴重的毒副作用,且化學性質穩定,殘留于動物源性食品中的氯霉素嚴重威脅著人類健康。因此,美國、歐盟、日本和我國等國家都將該類藥物列入禁止使用的獸藥名單中,并制定出相關的最高殘留限量(MRL)[2]。
對于出口蜂花粉,美國、歐盟、日本等國家都要求檢測其中的抗生素,如氯霉素、硝基咪唑類化合物等。目前氯霉素檢測主要集中在蜂蜜、蜂王漿、水產品、化妝品、肉制品等基質中[3-8],前處理主要采用乙酸乙酯、乙腈等有機試劑進行人工提取[9-11],操作步驟繁瑣,耗時。對于蜂花粉中氯霉素等抗生素檢測文獻報道較少[12-13]。因此,為了解決蜂花粉中氯霉素檢測技術問題,保證蜂花粉出口順暢,急需建立一套蜂花粉中氯霉素的檢測方法。
快速溶劑萃取法是近年新發展的一種前處理技術,在固體樣品中污染物、動物源性食品中獸藥殘留等的提取中有廣泛的應用,具有溶劑用量少、提取時間短和樣品提取自動化等優點[14-17]。超高效液相色譜-串聯質譜法采用1.8 μm填料色譜柱,同時采用三重四級桿串聯質譜儀進行檢測,具有分析速度快、分離度大、靈敏度高、定性準確等優點。因此,本研究采用快速溶劑萃取法進行萃取,硅膠固相萃取柱進行凈化,超高效液相色譜-串聯質譜法檢測,同位素內標法定量分析蜂花粉中氯霉素。
1 材料與方法
1.1材料與儀器
硅膠固相萃取柱500 mg,6 mL,上海安譜公司;C18固相萃取柱500 mg,6 mL,上海安譜公司;甲醇、乙酸乙酯色譜純,德國默克公司;乙酸乙酯、正己烷分析純,上海國藥;實驗用水超純水;氯霉素標準品德國Dr公司;氘代氯霉素標準品德國Dr公司;蜂花粉一部分為企業送檢樣品,一部分從浙江江山健康蜂業有限公司購買。
1260-6460液相色譜串聯四級桿質譜儀美國安捷倫科技有限公司;E-916快速溶劑萃取儀、R-210旋轉蒸發儀瑞士BUCHI公司;MS3 Digital漩渦振蕩器德國IKA公司;固相萃取裝置德國CNW公司;N-EVAPTM111氮吹儀美國organomation公司;BARNSTEAD NANOPURE超純水儀美國賽默飛科技中國有限公司。
1.2實驗方法
1.2.1標準溶液的配制和曲線繪制標準儲備液:分別精密稱取適量的氯霉素(CAP)標準品和氘代氯霉素(CAP-D5)標準品,用甲醇溶解并定容,配制成0.1 mg/mL的標準儲備液。該溶液在4℃的冰箱中保存。中間標準儲備液:分別準確吸取適量標準儲備液,用甲醇逐級稀釋成1.0 μg/mL中間標準儲備液。標準工作溶液:吸取不同體積的中間標準儲備液,用甲醇水溶液(體積比2∶8)稀釋成0.5、1.5、3.0、5.0、10.0 ng/mL的標準工作溶液,其中內標為5.0 ng/mL,現配現用。按照1.2.3和1.2.4建立的液相色譜條件和質譜條件進行測定,作待測物與其同位素內標物峰面積比對濃度比的標準曲線,求出直線回歸方程。
1.2.2樣品前處理方法準確稱取蜂花粉樣品2 g(精確至0.01 g)于150 mL燒杯中,加入100 μL濃度為100 ng/mL的氘代氯霉素內標溶液,加入2 g硅藻土,將樣品與硅藻土混合均勻,將混和好的樣品轉移至底部鋪有石英砂的萃取池中,上部再鋪一層石英砂,加蓋擰緊,將萃取池放入快速溶劑萃取儀中進行萃取,萃取液收集于240 mL收集瓶中,將萃取液轉移至雞心瓶中,45℃下旋蒸至近干。加入5 mL乙酸乙酯-正己烷(體積比2∶8)溶解殘渣,待凈化。
硅膠固相萃取柱用5 mL乙酸乙酯-正己烷(體積比2∶8)進行活化,將上述溶液轉移至萃取柱中,再加入5 mL乙酸乙酯-正己烷(體積比2∶8)洗滌雞心瓶,洗滌液轉移至萃取柱中,棄去流出液,用8 mL乙酸乙酯進行洗脫,接收全部洗脫液,45℃下氮氣吹干,加入1 mL甲醇水溶液(體積比2∶8),超聲1 min,渦旋混合1 min,過0.22 μm濾膜,待分析。
快速溶劑萃取儀萃取效率受萃取溶劑、溫度、壓力、循環次數等影響,因此,本研究對上述影響因素進行優化。在考察萃取溫度時,使用乙酸乙酯作為萃取溶劑、壓力90 bar、循環次數3次,實驗選取90、100、110、120℃4個溫度進行單因素實驗。在考察萃取壓力時,將萃取溫度設定為100℃、使用乙酸乙酯作為萃取溶劑、循環次數3次,實驗選取90、100、110、120 bar 4個萃取壓力進行單因素實驗。在萃取試劑為乙酸乙酯,萃取壓力100 bar,萃取溫度100℃,氮吹60 s,溶劑洗滌30 s條件下,分別選取靜態萃取循環次數2次和3次進行實驗。
對樣品凈化條件進行優化,取6支50 mL離心管,分別加入10 mL乙酸乙酯-正己烷(體積比2∶8)溶液,加入50 ng氯霉素標準溶液,將上述溶液轉移至已活化的硅膠固相萃取柱中,固相萃取柱用5 mL乙酸乙酯-正己烷(體積比2∶8)進行活化,再加入5 mL乙酸乙酯-正己烷(體積比2∶8)進行淋洗,棄去流出液,分別用5、6、7、8、9、10 mL乙酸乙酯進行洗脫,接收全部洗脫液,45℃下氮氣吹干,加入1 mL甲醇水溶液(體積比2∶8),超聲1 min,渦旋混合1 min,過0.22 μm濾膜,待分析。
1.2.3液相色譜條件色譜柱:Zorbax SB-C18(2.1 mm× 50 mm,1.8 μm);流動相:A相為超純水;B相為甲醇;柱溫40℃;流速:0.3 mL/min;進樣量10 μL;液相色譜流速及梯度洗脫見表1。

表1 液相色譜流速及梯度洗脫程序Table 1 HPLC flow rate and gradient elution program
1.2.4質譜參數離子源:電噴霧離子化源(ESI),負離子模式;干燥氣溫度:350℃;干燥氣流速:5 L/min;噴霧氣壓力:50 psi;鞘氣溫度:400℃;鞘氣流速:12 L/min;毛細管電壓:3000 V;多反應監測掃描(MRM)采集參數見表2。
2 結果與分析
2.1提取條件優化
2.1.1萃取溶劑選擇經過查閱大量文獻,提取氯霉素類抗生素的試劑主要有乙酸乙酯和乙腈[18-20]。但是乙腈的毒性較大,在對樣品進行高溫高壓提取過程中揮發出的乙腈會對人體產生毒害。而乙酸乙酯毒性相對較小,且在后續的濃縮過程中易蒸干,經過實驗驗證,乙酸乙酯能夠很好地將氯霉素從蜂花粉基質中提取出來,因此,本實驗選用乙酸乙酯為萃取溶劑。
2.1.2萃取溫度優化溫度是快速溶劑萃取的重要參數。隨著溫度的升高,溶劑的粘度下降,溶劑浸潤基質和溶解目標分析物的能力增強。但是隨著溫度的進一步提高,可能會促進部分抗生素的分解[21],導致回收率降低。經實驗優化,本實驗選擇100℃作為萃取溫度。
2.1.3萃取壓力優化 加壓的目的是使溶劑在高溫下仍然處于液態[22]。經實驗優化,當萃取壓力120 bar時,由于壓力較高,儀器易發生警報不能進行萃取。由圖2可知,萃取壓力為100 bar時萃取效率最高,本實驗選擇100 bar作為萃取壓力。
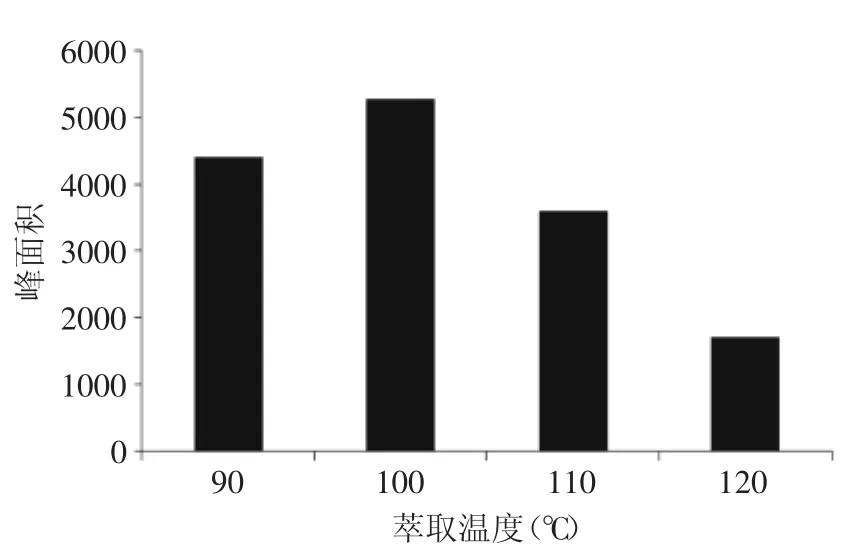
圖1 萃取溫度對氯霉素萃取效率的影響Fig.1 Effect of extraction temperature in ASE on extraction efficiency of chloramphenicol
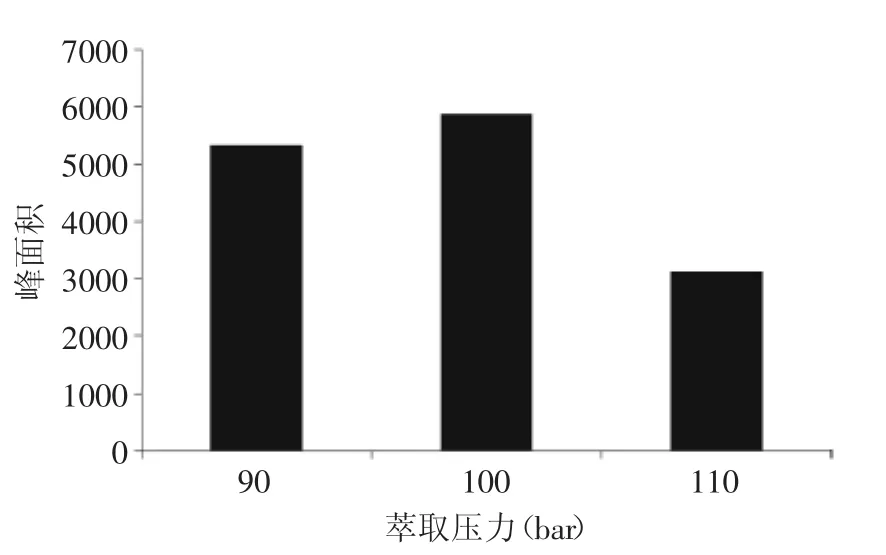
圖2 萃取壓力對氯霉素效率的影響Fig.2 Effect of extraction pressure in ASE on extraction efficiency of chloramphenicol
2.1.4循環次數優化靜態循環次數優化結果表明靜態萃取循環2次就基本能夠將氯霉素萃取完全。因此,本實驗選擇使用靜態萃取循環次數為2次。
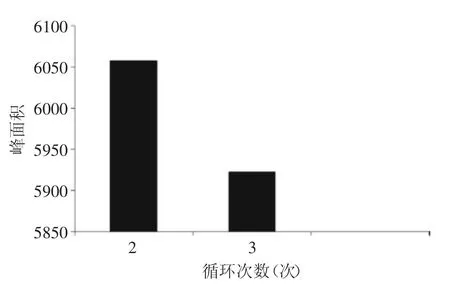
圖3 萃取循環次數對氯霉素萃取效率的影響Fig.3 Effect of extraction cycle index in ASE on extraction efficiency of chloramphenicol
2.2凈化條件優化
由于蜂花粉基質復雜,含有大量的天然色素、黃酮和纖維成分,在液質分析中存在嚴重的基質抑制作用。因此,需要通過一定的凈化方法去除干擾物質,降低基質抑制作用。本研究分別選取硅膠固相萃取柱和C18固相萃取柱進行比較,在凈化過程中發現C18固相萃取柱很容易被堵塞。而使用硅膠固相萃取柱沒有發生堵塞現象且凈化效果良好。因此,本研究選用硅膠固相萃取柱進行凈化。圖4為洗脫溶劑體積對氯霉素洗脫效果影響的比較。由圖4可以看出當洗脫體積為7和8 mL時,氯霉素的峰面積最大。當洗脫體積大于8 mL時,氯霉素峰面積反而減小。可能是由于洗脫體積增大,在后續氮吹等實驗過程中目標物損失較多。因此,本實驗室選擇使用8 mL洗脫液進行洗脫。
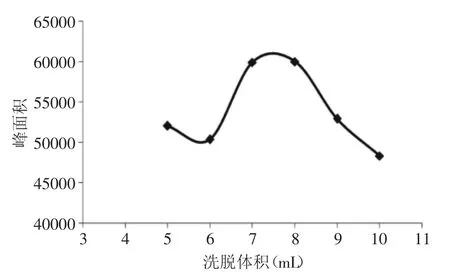
圖4 洗脫溶劑體積對洗脫效果影響的比較Fig.4 The comparison of elution effect with different eluting volume
2.3色譜條件優化
色譜柱是色譜分析技術的核心[23],本研究比較了Agilent Zorbax SB-C18(2.1 mm×50 mm,1.8 μm)和Agilent Eclipse XDB-C18(2.1 mm×100 mm,1.8 μm)2款色譜柱。2款色譜柱在實際的樣品檢測中均能將目標分析物和基質干擾物分離,且目標物峰型對稱、尖銳。Agilent Eclipse XDB-C18因其柱子較長產生的反壓比較大,且增加了分析時間,而Zorbax SB-C18具有更低的反壓、更快的分析速度,能夠大大的提高工作效率。因此,本實驗選擇Agilent Zorbax SB-C18作為液相色譜分析柱。
2.4質譜參數建立
本研究采用液相色譜進樣方式在負離子模式下對待測化合物的標準溶液進行母離子全掃描,確定準分子離子峰[M-1]-,再分別以待測化合物的準分子離子為母離子進行碎裂,進行二級子離子掃描,選擇2個特征子離子,選擇信噪比大、峰形好、干擾小的離子對作為定量離子對。優化后的質譜參數見表2,利用優化后的色譜和質譜條件對氯霉素進行分析。

表2 氯霉素液質測定條件Table 2 The optimized spectrometric parameters for chloramphenicol residue
2.5內標物選擇
應用液質聯用技術分析樣品中痕量獸藥殘留時,基質效應是時常遇到的現象,基質效應的出現可導致測試結果重復性差,定量結果難以保證[24]。同位素內標法是目前液質聯用分析中經常使用的一種定量技術,由于同位素內標物結構與目標分析物一致,受基質影響程度相同,在質譜中的碎裂行為相似[25]。因此,同位素內標法可有效的降低基質效應的影響,提高定量的準確性。本文選用氘代氯霉素(CAP-D5)作為內標物。
2.6方法的線性關系、檢出限和定量限
在本研究所確定的條件下,以目標分析物與其同位素內標物濃度比值為橫坐標x,以目標分析物與同位素內標物峰面積比值為縱坐標y,繪制氯霉素標準溶液工作曲線,氯霉素在0.5~10 ng/mL范圍內呈良好的線性關系。該方法的檢出限和定量限由空白樣品添加回收實驗獲得,以3倍信噪比(RSN)為檢出限、10倍信噪比(RSN)為定量限,氯霉素的檢出限和定量限分別為0.1 μg/kg和0.3 μg/kg。

表3 氯霉素的線性方程及線性范圍Table 3 Linear ranges and regression equations of Chloramphenicol residue
2.7精密度和回收率
為測定本方法的精密度和回收率,實驗選用空白蜂花粉為樣品基質,添加不同量的氯霉素標準溶液,進行加標回收實驗,加標水平為0.3、0.6、1.0 μg/kg,每個添加水平均做6個平行樣,其結果見表4。由表4可以看出,氯霉素的加標回收率在88.3%~110.0%之間,相對標準偏差在5.3%~8.9%之間,能夠滿足分析方法的要求。

表4 回收率與精密度實驗結果(n=6)Table 4 Mean recovery rates and precision for 6 replicate determinations(n=6)
2.8實際樣品分析
將本研究建立的方法編寫成實驗室的標準操作規程,應用于實驗室的檢測工作。按照1.2.2的前處理方法,對送檢和購買的6批次蜂花粉樣品進行檢測,雖然有個別樣品中有氯霉素峰,但是都小于方法檢出限。其中有峰的兩個樣品,結果分別為0.08 μg/kg和0.05 μg/kg。下一步將繼續使用該方法檢測企業委托送檢的樣品,積累更多的數據。
3 結論
本研究建立了蜂花粉中氯霉素殘留檢測的超高效液相色譜-串聯質譜(UPLC-MS/MS)分析方法。通過對液相色譜、質譜、快速溶劑萃取儀、硅膠固相萃取柱條件進行優化。經方法學驗證,方法線性范圍0.5~10 ng/mL,相關系數為R2=0.9936,回收率在88.3%~110.0%之間,相對標準偏差在5.3%~8.9%之間,檢出限為0.1 μg/kg。本研究與文獻[12]中的方法相比,在萃取環節采用了快速溶劑萃取儀進行萃取,減少了人工操作,自動化水平大大提高。在凈化過程中,文獻[12]使用2種固相萃取柱進行凈化,本研究僅使用1種固相萃取柱進行凈化,大大降低了時間和物力成本。本研究使用同位素內標法進行定量,方法的靈敏度、準確度和精密度均能滿足獸藥殘留分析方法的要求。
[1]宋巍巍,柴春彥,劉國艷,等.動物源性食品中氯霉素檢測方法的研究進展[J].畜牧與獸醫,2007,39(4):54-56.
[2]吳永寧,周宗燦,王緒卿.現代食品安全科學[M].北京:化學工業出版社,2003:153-182.
[3]沈崇鈺,丁濤,陳惠蘭.高效液相色譜-電噴霧多級質譜聯用測定蜂產品中氯霉素殘留[J].檢驗檢疫科學,2003,13(6):26-28.
[4]孟憲雙,馬強,李晶瑞,等.高效液相色譜-串聯質譜法同時測定祛痘化妝品中的3種氯霉素類抗生素[J].分析實驗室,2014,33(3):332-336.
[5]王清,王國民,郗存顯,等.復合免疫親和柱凈化-液相色譜-串聯質譜法同時測定動物源性食品中6種玉米赤霉醇類化合物和氯霉素殘留量[J].色譜,2014,32(6):640-646.
[6]謝世紅,謝世濤,孟霞,等.液相質譜法同時測定水產品中氯霉素和硝基呋喃類代謝物殘留量的研究[J].現代科學儀器,2014,3:232-238.
[7]王浩,楊紅梅,郭啟雷,等.液相色譜-串聯質譜法同時測定嬰幼兒配方乳粉中氯霉素、三聚氰胺、甲硝唑和洛硝噠唑[J].分析化學研究簡報,2013,41(2):283-287.
[8]高潔.氣相色譜-串聯質譜法同時測定肉制品中氯霉素、甲砜霉素殘留[J].中國釀造,2013,41(2):283-287.
[9]王立琦,賀利民,曾振靈,等.液相-串聯質譜檢測獸藥殘留中的基質效應研究進展[J].質譜學報,2011,32(6):321-332.
[10]彭濤,李淑娟,侯曉剛,等.高效液相色譜/串聯質譜法同時測定蝦中氯霉素、甲砜霉素和氟甲砜霉素殘留量[J].分析化學,2005,33(4):463-466.
[11]Takino M,Daishima S,Nakahara T.Determination of chloramphenicol residues in fish meats by liquid chromatographyatmospheric[J].Journal of Chromatography:A,2003,1011:67-75.
[12]Fujita K,Ito H,Nakamuar M,et al.Determination of chloramphenicol residues in bee pollen by liquid chromatography/ tandem mass spectrometry[J].J AOAC Int,2008,91(5):1103-1109.
[13]楊雯筌,辛志宏,殷耀,等.LC-MS-MS檢測蜂花粉中硝基呋喃類代謝物[J].食品科學,2013,34:183-190.
[14]左艷麗,孫漢文,魏立靜.快速溶劑萃取-毛細管電泳法測定土壤和底泥中磺胺類藥物殘留[J].分析實驗室,2012,31(2):62-66.
[15]楊洪生,孟勇,張美琴,等.快速溶劑萃取-超高效液相色譜-串聯質譜法同時測定水產品中氯霉素和氟苯尼考[J].理化檢驗-化學分冊,2012,48:1353-1356.
[16]Carabias-Martfnez R,Rodriguez-Gonzalo E,Revilla-Ruiz P,et al.Pressurized liquid extraction in the analysis of food and biological samples[J].Journal of Chromatography:A,2005,1089(1/2):1-17.
[17]Mendiola J A,Herrero M,Cifuentes A,et al.Use of compressed fluids for sample preparation:Food applications[J]. Journal of Chromatography:A,2007,1152(1/2):234-246.
[18]馮民,魏云計,朱臻怡,等.高效液相色譜-串聯質譜法同時測定飼料中氯霉素、甲砜霉素與氟甲砜霉素殘留量[J].分析測試學報,2013,32(1):117-121.
[19]羅輝泰,黃曉蘭,吳慧勤,等.QuEChERS/液相色譜-串聯質譜法同時測定魚肉中30種激素類及氯霉素類藥物殘留[J].分析測試學報,2011,30(12):1329-1337.
[20]胡雪,李翠枝,劉麗君,等.UPLC-MS-MS法測定飼料、原料乳及乳制品中3種氯霉素類藥物殘留[J].食品科學,2014,35(16):110-113.
[21]Carretero V,Blasco C,Pico Y,et al.Multi-class determination of antimicrobials in meat by pressurized liquid extraction and liquid chromatography-tandem mass spectrometry[J].Journal of Chromatography:A,2008,1209(1/2):162-173.
[22]劉靜.ASE快速溶劑萃取-解決固體、半固體樣品前處理的新技術[J].現代科學儀器,2002(3):59-61.
[23]郝昀,李揮,孫漢文,等.加速溶劑萃取在動物源食品農獸藥殘留分析中的應用進展[J].河北大學學報:自然科學版,2012,32(4):434-448.
[24]石先哲,李攻科,梁振,等.色譜研究最新進展[J].化學通報,2014,77(7):720-730.
[25]向平,沈敏,卓先義.液相色譜-質譜分析中的基質效應[J].分析測試學報,2009,28(6):753-756.
Determination of chloramphenicol residue in bee pollen by accelerated solvent extraction-ultra performance liquid chromatography-tandem mass spectrometry
ZHU Zi-tong,LEI Mei-kang,PENG Fang,LI Dong,CHEN Yu-jiao,ZHANG Ying-jun
(Comprehensive Technology Center of Quzhou Entry-Exit Inspection and Quarantine Bureau,Quzhou 324002,China)
A method for analysis of chloramphenicol residue in bee pollen was developed.Sample was extracted using accelerated solvent extraction with ethyl acetate as extraction reagent and purified by Silica solid phase. Mass spectrometer was operated in the negative ion mode using select reaction monitoring.Isotope internal standard was used for quantitative analysis.The linearity of the calibration curve was from 0.5 to 10 ng/mL with correlation coefficient was 0.9936.Recovery rates for chloramphenicol in a negative bee pollen sample spiked at three levels were between 88.3%and 110.0%,with RSD range of 5.3%~8.9%.The limit of detection in the method was 0.1 μg/kg.This method proved to be of high efficiency,small amount of reagent,good selectivity and high sensitivity.
bee pollen;accelerated solvent extraction(ASE);ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);chloramphenicol(CAP)
TS201.1
A
1002-0306(2015)20-0068-05
10.13386/j.issn1002-0306.2015.20.005
2015-01-20
祝子銅(1985-),男,碩士,工程師,研究方向:食品及動物源性產品中獸藥殘留檢測,E-mail:zztzzt1124@163.com。
