槲皮素在分子印跡聚合物修飾碳糊電極上的電化學行為及測定
2015-12-17 00:36:56張旭紅翟海云潘育方
中國測試 2015年11期
關鍵詞:實驗
張旭紅,周 清,翟海云,潘育方
(1.廣東藥學院中藥學院,廣東 廣州 510006;2.廣東藥學院藥科學院,廣東 廣州 510006)
0 引 言
槲皮素(Quercetin)廣泛存在于植物中,具有抗氧化及清除自由基、保護心血管系統以及抗病毒等藥理活性[1-3]。關于槲皮素含量的測定方法已有較多相關報道,主要有紫外分光光度法,反相高效液相色譜法等,也有相關文獻采用電化學方法進行含量測定。現將相關測試方法的一些特點在表1中列出。
本實驗采用操作成本低,選擇性、靈敏度均較高的分子印跡聚合物修飾碳糊電極法測定藥物中的槲皮素含量。單純的碳糊電極(CPE)作用有限,但可以通過電極修飾的方法使碳糊電極具有一定的功能,即化學修飾碳糊電極。而分子印跡聚合物(MIP),具有較好的通用性和立體專一識別性,已廣泛應用于色譜分離、生物傳感器以及酶的模擬和催化合成等領域[8-9]。本實驗以經過分子印跡聚合物修飾的碳糊電極(MIP-CPE)作為工作電極,測定槲皮素的電化學行為,并且應用于膠囊中槲皮素含量測定。結果顯示該方法穩定可靠,檢出限低,檢測方便,易于推廣。
1 實驗部分
1.1 儀器與試劑
Ingsens-1000系列掌上型電化學工作站(盈思儀器(廣州)有限公司);分子印跡聚合物修飾碳糊電極(自制)為工作電極;Ag-AgCl(飽和KCl)電極為參比電極、鉑電極為對電極(上海辰華儀器有限公司);pHS-25型酸度計(上海雷磁新涇儀器有限公司)。
槲皮素(中國藥品生物制品檢定所),槲皮素維生素C復合膠囊(普瑞登健康食品公司),甲基丙烯酸(MAA,AR,成都市科龍化學試劑廠),乙二醇二甲基丙烯酸酯(EGDMA,AR,阿拉丁試劑有限公司),偶氮二異丁腈(AIBN,AR,天津市科密歐化學試劑有限公司),石墨粉(GR,國藥集團化學試劑有限公司),液體石蠟(AR,成都市科龍化學試劑廠)。其余試劑均為分析純,實驗用水為蒸餾水。
1.2 分子印跡聚合物的制備
稱取0.3022g槲皮素溶于100mL甲醇中,印跡分子、MAA、EGDMA 的比例采用經典的摩爾比 1∶4∶20,按比例加入功能單體(MAA)0.34mL、交聯劑(EGDMA)3.77mL,最后加入引發劑(AIBN)0.09g充分混勻后,向混合液中通氮氣10min,蠟封后置于恒溫水浴箱,60℃聚合24h。聚合產物取出冷卻至室溫,用醋酸-甲醇(V∶V=1∶9)溶液淋洗至洗脫液中檢測不到槲皮素分子,再用甲醇洗去過量的醋酸,于烘箱中40℃干燥即得槲皮素分子印跡聚合物。
1.3 修飾碳糊電極制作和活化
石墨粉和印跡聚合物質量比為4∶1,按照m(石墨+聚合物)∶m(石蠟)=4∶1[10]混合均勻后,裝入半徑為2.0mm的玻璃管中壓緊,用銅絲線做導線,引出后固定電極,將電極表面在平滑的白紙上磨平拋光,備用。在pH 3.6的HAc-NaAc緩沖液中以100 mV/s的掃速于0~0.6V循環掃描20次,以活化電極表面。
非分子印跡聚合物修飾碳糊電極(NIP-CPE)的制作除了不加槲皮素模板因子以外,其余與MIP-CPE制作步驟相同。
1.4 槲皮素標準溶液配制
0.001 mol/L槲皮素標準溶液的配制:準確稱取0.0302g槲皮素用少量的乙醇溶解后用乙醇定容于100mL容量瓶中,然后置于冰箱內冷藏,待使用時用蒸餾水稀釋至所需濃度。
1.5 樣品前處理
稱量并計算平均每粒槲皮素維生素C復合膠囊(Puritian’s Pride Inc,標示量為 250mg/粒)的質量。為防止維生素C對檢測結果的干擾,取4粒膠囊,傾取內容物溶于200 mL的去離子水中,過濾,取濾渣干燥,并稱取濾渣1/2的質量,約含槲皮素125 mg,裝在50 mL錐形瓶中,加甲醇約20 mL,超聲30 min后,轉移至50 mL容量瓶中,甲醇定容,微孔濾膜過濾,取續濾液1 mL于25 mL容量瓶中,并用甲醇定容,備用。

表1 槲皮素含量測定方法特點
1.6 電化學測試
取1.4、1.5項下的溶液適量,用pH 3.6的HAc-NaAc緩沖溶液稀釋至刻度,搖勻即為樣品溶液。將樣品溶液移入小燒杯中,插入三電極系統,掃速100 mV/s從0 V掃描至0.6 V,富集150 s,記錄其在0.34V附近的峰電流。
2 結果與討論
2.1 槲皮素分子印跡效應
在物質量濃度為1.0×10-4mol/L槲皮素溶液中,以100mV/s的掃速在0~0.6V間掃描。結果如圖1所示,分子印跡聚合物修飾碳糊電極有良好的響應效果,而非分子印跡聚合物修飾碳糊電極與裸碳糊電極對槲皮素響應均較小。圖1中a是槲皮素在MIP-CPE上的連續掃描循環伏安曲線,陽極掃描有峰,其峰電位在0.338V,而反向掃描過程中,亦有觀察到相應的還原峰,其峰電位為0.276V,說明槲皮素在MIP-CPE上的氧化是可逆的。
可能是由于槲皮素印跡聚合物具有分子印跡效應,槲皮素分子可通過印跡孔穴擴散至分子印跡聚合物修飾碳糊電極表面,發生電化學氧化反應,與印跡識別位點之間的親和性增加。該結果表明槲皮素分子印跡聚合物對槲皮素模板分子識別吸附能力增強。

圖1 槲皮素的分子印跡效應
2.2 測試條件的確定
2.2.1 聚合物修飾碳糊電極的組成
碳糊電極中,石墨-液體石蠟比例為4∶1時電極的效果最好,峰電流最大,峰型相對好。因此,在制備分子印跡聚合物修飾碳糊電極時,以m(石墨+聚合物)∶m(石蠟)=4∶1制作工作電極,比較石墨粉與分子印跡聚合物不同質量比(90∶10,80∶20,70∶30,60∶40,55∶45)對掃描結果的影響。活化后,分別在5.0×10-5mol/L槲皮素溶液中,以100mV/s的掃速從0V掃描至0.6V,掃描結果見圖2,當石墨粉與分子印跡聚合物的比例為80∶20時,有較強的信號及較好的峰形,對槲皮素的識別能力明顯增強。故本次實驗以m(石墨+聚合物)∶m(石蠟)=4∶1,其中石墨粉與分子印跡聚合物的比例為80∶20,制作實驗的工作電極。

圖2 分子印跡聚合物修飾碳糊電極組成對峰電流的影響
2.2.2 緩沖溶液的選擇
在 NaH2PO4-NaOH、Na2SO4-NaOH、B.R、H3PO4、HAc-NaAc、HNO3、H2SO4、HCl等緩沖溶液中分別進行試驗。結果發現在強堿和中性溶液中氧化峰均不明顯,在弱酸中氧化峰較好。在HAc-NaAc緩沖溶液中波形最好,峰電流最大,穩定性好,故選擇HAc-NaAc緩沖溶液為底液。
2.2.3 pH值對峰電流的影響
當槲皮素物質量濃度為1.0×10-4mol/L時,研究峰電流隨pH值的變化規律,結果如圖3所示,峰電流隨著pH值增大呈現先增大后減小再增大的變化規律,但增大后的響應值不如較低pH值時的峰電流值大。由圖可知,在pH 3.6附近的峰電流相對較大且穩定,峰形較好,故選擇pH 3.6的HAc-NaAc溶液為底液。
2.2.4 攪拌富集時間對峰電流的影響
圖4是在2個不同槲皮素濃度下測定攪拌富集時間對峰電流的影響。當物質量濃度為1.0×10-4mol/L時,峰電流在110s出現最大值;物質量濃度為1.0×10-5mol/L時,峰電流在150s出現最大值。由圖可知,峰電流隨攪拌富集時間增加先增大后減小。濃度不同的槲皮素峰電流值達到最大值所需要的時間不同。濃度越小,達到最大值所需要的時間越長;濃度越大,達到最大值所需要的時間越短。為了使不同濃度下富集時間更合理,實驗時選擇150s的富集時間。

圖3 pH值對峰電流的影響

圖4 攪拌富集時間對峰電流的影響
2.2.5 掃描速率ν對峰電流的影響
用循環掃描伏安法研究了掃描速率ν對峰電流的影響,結果發現槲皮素的氧化峰電流與掃描速率在10~400mV/s之間呈現出一定的線性關系,如圖5所示,線性方程為 y=0.0049x-0.0241(r=0.992),x 為掃描速率(mV/s),y為峰電流(μA)。實驗發現峰電流隨著掃描速率增大而增大,峰電位向高電位方向移動,但峰型會隨著掃描速率增大而變差。為了獲得較好的峰型,實驗中掃描速率選擇為100mV/s。不同掃描速率下,峰形的變化情況如圖6所示。

圖5 掃描速率與峰電流的線性關系
2.3 線性范圍與檢出限
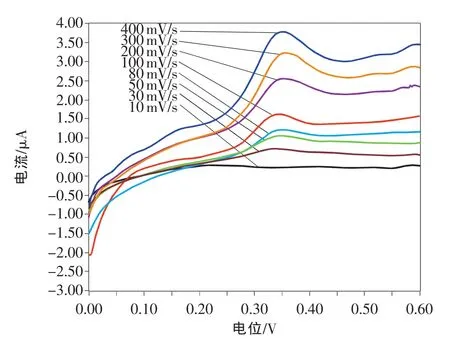
圖6 不同掃描速率對峰形的影響

圖7 槲皮素濃度與峰電流的線性關系

圖8 標準曲線的循環伏安圖
在最佳實驗條件下,槲皮素在1.25×10-6~4.0×10-5mol/L范圍內與峰電流呈良好線性關系(見圖7),回歸方程為:y=0.0918x+0.6408,r=0.9950,濃度梯度分別為 1.25×10-6,2.5×10-6,5.0×10-6,1.0×10-5,2.0×10-5,4.0×10-5mol/L)。槲皮素不同濃度標準溶液在MIPCPE上的循環伏安圖如圖8所示,隨著槲皮素標準溶液濃度增大,響應值峰電流依次增大。
固定槲皮素物質量濃度在5.0×10-6mol/L,測定響應峰電流(n=12),計算檢出限為 4.72×10-8mol/L。結果顯示,槲皮素分子印跡聚合物修飾碳糊電極線性范圍較寬,且具有較高的靈敏度。
2.4 樣品分析
分子印跡聚合物修飾碳糊電極測定槲皮素維生素C復合膠囊中槲皮素含量,按1.5.項下方法處理樣品,最終測得復合膠囊中槲皮素平均含量為每粒251.8mg(n=3,RSD=3.0%)。實驗表明經處理后膠囊中的共存物對測定的干擾較少。回收率測定結果見表2。

表2 膠囊中槲皮素含量的測定結果及回收率(n=3)
3 結束語
分子印跡聚合物特異性識別能力較高,能明顯增強槲皮素在電極上的響應信號及其抗干擾的能力。與紫外分光光度法、高效液相色譜法以及其他相關電化學方法相比,分子印跡聚合物修飾碳糊電極法具有選擇性高、檢出限低、分析時間較短、分析儀器體積小巧、安全性相對較高、易于推廣等優勢。
本文研究了槲皮素在分子印跡聚合物修飾碳糊電極上的伏安行為,建立了利用該修飾電極測定槲皮素的電化學分析方法,并測定了槲皮素維生素C復合膠囊中的槲皮素含量,結果滿意。修飾電極是自制的,廉價、無毒,制作過程方便簡單,電極表面容易更新而且靈敏度相對較高;電極的表面積小,對樣品的消耗量少,實驗操作方便,所測樣品處理要求簡單,易于推廣到藥理和臨床醫學的研究。
[1] 劉娜,王文平,康倩,等.銀杏葉黃酮類成分體外及經腸吸收后抗氧化活性比較[J].天津中醫藥,2015,32(1):42-45.
[2] 湯喜蘭,劉建勛,李澎,等.山柰酚和槲皮素對乳鼠心肌缺氧復氧及過氧化損傷的保護作用[J].中藥藥理與臨床,2012,28(1):56-59.
[3] 張丹丹,方建國,陳娟娟,等.連翹及其主要有效成分槲皮素體外抗人巨細胞病毒的實驗研究[J].中國中藥雜志,2010,35(8):1055-1059.
[4] 趙超,陳華國,靳鳳云,等.槐枝中總黃酮的含量測定[J].光譜實驗室,2010,27(1):188-191.
[5] 賓婕,劉潔,陳克嶙,等.不同來源苦蕎中蘆丁和槲皮素的含量測定[J].現代食品科技,2011,27(1):117-119.
[6] 朱玲艷,王宗花,陳小印,等.聚吡咯/碳納米管分子印跡修飾電極對槲皮素的選擇性測定[J].分析測試學報,2011,30(1):18-23.
[7] 鄧光輝,王士偉,王輝,等.毛細管電泳安培法測定田基黃中的蘆丁與槲皮素[J].分析試驗室,2014,33(4):424-427.
[8] 賀倩倩,張高奎,賀利民.分子印跡聚合物及其在藥物分離中的應用研究進展[J].中國獸藥雜志,2011,45(9):48-55.
[9] 伍華,朱佳,段景峰,等.化學修飾碳糊電極的研究新進展[J].應用化工,2012,41(5):880-883.
[10]龍寧,朱明芳,嚴志紅,等.陽極吸附伏安法測定鹽酸莫西沙星[J].理化檢驗:化學分冊,2012(48):37-39.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
