柱前衍生-反相高效液相色譜法檢測富硒米曲霉中有機硒形態
2015-12-20 08:54:08李紅衛王開萍唐正江劉國慶
食品科學 2015年22期
李紅衛,王開萍,吳 娛,唐正江,湯 飛,劉國慶,2,*
(1.合肥工業大學生物與食品工程學院,安徽 合肥 230009;2.安徽省皖江禽產業研究院,安徽 宣城 242000)
柱前衍生-反相高效液相色譜法檢測富硒米曲霉中有機硒形態
李紅衛1,王開萍1,吳 娛1,唐正江1,湯 飛1,劉國慶1,2,*
(1.合肥工業大學生物與食品工程學院,安徽 合肥 230009;2.安徽省皖江禽產業研究院,安徽 宣城 242000)
建立高效液相色譜法測定富硒米曲霉中有機硒形態的分析方法。采用6 mol/L HCl溶液,50 ℃水解富硒米曲霉48 h,提取樣品中的有機硒,4-氯-3,5-二硝基三氟甲苯柱前衍生,C18反相色譜柱(4.6 mm×250 mm,5 μm)分離,以乙腈-水(1∶1,V/V)和pH 6.8乙酸-乙酸鈉緩沖液為流動相進行梯度洗脫,紫外檢測波長:240 nm,測得富硒米曲霉中有機硒化合物主要是硒代胱氨酸,檢出限(RSN=3)為0.680 mg/L,定量限(RSN=10)為2.244 mg/L,線性范圍為5.0~100 mg/L,加標回收率為94.8%~105.8%,硒代胱氨酸的含量為1.25 mg/g。本方法簡單、靈敏、重復性好,可適用于富硒米曲霉及相關產品中有機硒形態的檢測分析。
富硒米曲霉;有機硒;硒代氨基酸;柱前衍生-反相高效液相色譜法
硒是人和許多生物體必需的微量元素,對生物體的健康具有重大意義。市面上出現了許多富硒產品,如酵母硒、植物硒以及一些富硒食用菌等,其品質的高低不僅取決于硒的含量,還與硒的形態有著密切關聯。研究[1-2]表明:無機硒生物活性、利用率較低,不易被腸道吸收,還具有一定毒性,攝入過量會導致急性、慢性中毒;與無機硒相比,有機硒是經生物轉換而得,具有毒性小、生物利用度高的特點,并且硒的所有生物學功能均是通過硒蛋白來調控和實現的,因此有機硒相較于無機硒對生物體來說,更為重要。
米曲霉是一類產復合酶的菌株,除產蛋白酶外,還可產淀粉酶、糖化酶、纖維素酶、植酸酶等,被廣泛應用于食品、飼料、生產曲酸、釀酒等發酵工業,并已被安全地應用1 000多年。潘艷[3]利用米曲霉作為富硒載體,在其培養基中加入無機硒,利用生物轉化將難吸收的無機硒源轉化為易吸收的有機硒源[4],發酵生產富硒米曲霉,測得搖瓶發酵的富硒量達2.512 mg/g,分批發酵的富硒量更是高達3.3 mg/g,遠高于富硒酵母的1.0~2.0 mg/g硒含量。現階段關于富硒酵母、植物硒和一些其他富硒產品中硒形態[5-7]的研究已經較為完善,但富硒米曲霉中硒形態的研究這一領域仍然比較缺乏,因此建立一種分析富硒米曲霉中硒形態及含量的方法,不僅可以為該領域的研究提供一定的理論依據,也對評價其營養價值具有重要意義。與此同時,還能夠對開發高產、優質的富硒米曲霉保健品起促進作用。
近些年來,國內很多研究者對富硒樣品中硒的形態做了大量研究。陳尚衛等[8]采用酶解提取法提取,氯甲酸乙酯衍生,氣相色譜-串聯質譜(gas chromatographytandem mass spectrometry,GC-MS-MS)聯用法選擇離子模式對富硒酵母中的硒代蛋氨酸進行檢測;劉揚等[9]采用蛋白酶提取富硒酵母中的有機硒,高效液相色譜-電感耦合等離子體質譜(high performance liquid chromatographyinductively coupled plasma mass spectrometry,HPLC-ΙCPMS)聯用法檢測其有機硒的形態;Deng Biyang等[10]采用稀酸提取,毛細管電泳-電化學發光(capillary electrophoresis coupled with electrochemiluminescence,CE-ECL)法檢測富硒酵母中硒代蛋氨酸;仲娜[11]采用微波消解法提取市場上出售的富硒大米、富硒茶葉、富硒螺旋藻、富硒大蔥、富硒雞蛋中的有機硒,HPLC-ΙCPMS測其硒形態。以上這些方法,HPLC-ΙCP-MS聯用儀器昂貴,普及度不高,樣品預處理繁瑣,分析成本較高;CE-ECL方法中電泳的重現性不高,影響定性定量結果。考慮到以上這些因素,并且目前關于富硒米曲霉中硒形態的研究也較少,本實驗針對其硒形態的分析建立一種方法,采用酸水解法進行樣品提取,并用4-氯-3,5-二硝基三氟甲苯(4-chlorine-3,5-dinitrobenzotrifl uoride,CNBF)對樣品提取液進行衍生,衍生過后直接運用反相高效液相色譜法對樣品中的有機硒化合物形態和含量進行分析。
1 材料與方法
1.1 材料與試劑
富硒米曲霉(硒含量2.325 mg/g) 實驗室自制。
標準樣品:硒代甲硫氨酸(SeMet,98%,CAS:3211-76-5)、硒代胱氨酸(SeCys2,97%,CAS:29621-88-3) 美國Sigma公司;衍生劑:CNBF 美國阿拉丁公司;超純水、pH 7.0磷酸緩沖液、pH 9.0硼砂-硼酸緩沖液、pH 6.8乙酸-乙酸鈉(0.05 mol/L)緩沖液、甲醇(色譜純)、乙腈(色譜純)、0.08 mol/L NaOH、 6 mol/L HCl溶液。
1.2 儀器與設備
2695高效液相色譜儀配紫外檢測器 美國Waters公司;超微電子天平 德國Sartorius公司;SE812氮吹儀北京帥恩科技有限公司;DHG-9140A電熱恒溫鼓風干燥箱 鞏義市予華儀器有限責任公司;恒溫水浴鍋國華電器有限公司;HC-3518型高速離心機 安徽中科中佳科學儀器有限公司;PHS-3C型精度酸度計 上海大普儀器有限公司;WH-861型渦旋振蕩器 太倉市科教儀器廠;超聲波清洗器 昆山超聲波儀器有限公司。1.3 方法
1.3.1 標準儲備溶液的配制
分別稱取適量的SeMet和SeCys2標準品,用超純水制0.1mg/mL的標準品儲備液,溶液置于4 ℃冰箱中保存備用,保存期限一個月。
1.3.2 樣品預處理
準確稱取0.3g經液氮研磨的富硒米曲霉粉末4 份置于10 mL的離心管中,加入5 mL 6 mol/L HCl溶液,用氮氣吹10 min后封口。一份在110℃條件下水解24 h,另外3 份在50℃條件下分別水解24、48、72 h。離心15 min后取上清液,用超純水定容至100 mL備用。
1.3.3 樣品的衍生
取3 mL樣品溶液,加入2 mL pH 9.0硼砂-硼酸緩沖液和100 μmol/mL的CBNF衍生劑2 mL,封口,在65 ℃水浴條件下衍生40 min,反應結束后取出冷卻,放置至室溫,振蕩,用pH 7.0磷酸緩沖液定容至10 mL后振蕩30 s,再靜置10 min。用0.22 μm有機過濾器過濾至上機小瓶,待用。
分別取0.1 mL的SeMet和SeCys2標準溶液儲備液按照如上方法進行衍生。其反應機理[12]見圖1。

圖1 SeMet與CNBF(a)、SeCys2與CNBF(b)和CNBF自身水解(c)反應機理圖Fig.1 (a) Reaction scheme of CNBF with amine groups on SeMet; (b) Reaction scheme of CNBF with amine groups in SeCys2; and (c) hydrolysis of CNBF reagent
1.3.4 色譜條件
色譜柱:We l c h U l t i m a t e C18反相色譜柱(4.6 mm×250 mm,5 μm);流動相A:乙腈-水(1∶1,V/V);流動相B:0.05 mol/L,pH 6.8乙酸-乙酸鈉緩沖液;柱溫:31 ℃;檢測波長:240 nm;流速1.2 mL/min;梯度洗脫:0~25 min,65% A、35% B;25~30 min,75% A、25% B;30~34 min,90% A、10% B;34~40 min,100% A、0% B。
2 結果與分析
2.1 硒代氨基酸提取方法的選擇
富硒米曲霉中硒代氨基酸是有機硒的主要形式,而有機硒主要存在于蛋白中,故采用水解蛋白的方式提取富硒米曲霉中的硒代氨基酸。目前蛋白水解的方式主要以酸水解法為主,但是普通的酸水解條件為110 ℃條件下水解24 h,在該方法中甲硫氨酸、胱氨酸/半胱氨酸常常會遭到破壞,易造成損失[13]。郝素娥等[14]采用6 mol/L的HCl溶液,50℃水解50 h的方法提取富硒酵母中的硒化合物,有機硒提取率達到61.3%;唐瑩瑩等[15]用6 mol/L的HCl溶液提取富硒大米中的硒化合物,提取時間分別為24、48 h,相對應的提取溫度為110 ℃和50 ℃。結果顯示50 ℃條件下提取48 h所得的硒化合物含量高于110 ℃條件下提取24 h的量。因此本實驗預建立一種條件溫和且操作簡單的前處理水解法,即降低水解溫度對富硒米曲霉中的硒代氨基酸進行提取,并且比較了不同水解時間條件下,有機硒的提取量,其結果見表1。

表1 富硒米曲霉中硒化合物的測定Table1 The contents of selenium compounds in selenium-enriched Aspergillus oryyzzaaee
由表1可知,4 種水解方法相比較而言,50 ℃、24 h的水解條件下,提取的SeCys2的量最低,僅為1.12 mg/g,推測可能是由于蛋白未水解徹底導致;50 ℃、72 h條件下的最高,為1.27 mg/g,比110 ℃水解24 h的1.18 mg/g高7.6%,并且略高于50℃、48 h條件下的1.25 mg/g,SeMet均未檢出。綜合考慮實驗條件與實驗時間,選取反應條件較溫和,反應時間較短的50℃、48 h為本實驗的水解條件。
2.2 衍生條件的優化
2.2.1 衍生劑的選擇
SeMet、SeCys2及大部分氨基酸本身并不能產生紫外吸收,必須經過衍生化反應,產生具有紫外吸收的衍生物才能被檢測。目前國內外應用較廣泛的衍生劑有鄰苯二甲醛(o-phthalaldehyde,OPA)[16-17]、異硫氰酸苯酯(phenyl isothiocyanate,PΙTC)[18]、2,4-二硝基氟苯(dinitrofluorobenzene,DNFB)[19]、丹酰氯(Dansyl-Cl)[20]、6-氨基喹啉基-N-羥基-琥珀酰亞胺基甲酸酯(6-aminoquinolyl-N-hydroxysuccinimidyl carbamate,AQC)[21-22]、9-芴甲基氯甲酸酯[23-24]和CNBF等。
雖然衍生劑的種類很多,但沒有哪一種衍生劑能夠完全適合所有氨基酸的分析[25-27]。例如,OPA衍生法具有衍生步驟簡單、反應速度快、剩余試劑不干擾測定等優點,缺點是只能與一級氨基酸反應,二級氨基酸不能與其直接反;Dansyl-Cl法可同時檢測包括亞氨基酸在內的所有氨基酸,缺點是流動相中的雜質和衍生副產物對測定有干擾;DNFB法的優點有衍生產物穩定、操作簡便、成本低等,衍生產物由于硝基苯環的引入,有較強的紫外吸收,因此具有較高的靈敏度,缺點是衍生過程中產生的副產物干擾測定,限制了DNFB在氨基酸分析中的應用;PΙTC法可同時測定伯氨酸和仲氨酸,衍生產物單一、穩定,缺點是PΙTC毒性大,且極易損耗色譜柱壽命。CNBF衍生條件為65 ℃條件下衍生40 min,與以上衍生劑相比具有反應條件溫和、衍生時間短、成本低等優勢,因此本實驗采用CNBF做衍生劑進行硒代氨基酸的檢測。
2.2.2 衍生劑用量對衍生效率的影響
為使衍生反應完全,一般需要加入過量的衍生劑。實驗了不同用量衍生劑對衍生效率的影響,當反應溫度65 ℃、反應時間40 min時,隨著衍生劑用量的增大,色譜峰面積增大,一般當衍生劑用量大于20 倍理論量時,峰面積不在變化,衍生反應接近完全。本法選擇3.0 mL樣品溶液加入2 mL 100 mmol/L的CNBF衍生劑進行衍生反應,約相當于硒代氨基酸標準品量為0.1 mg/mL時的40 倍(考慮到CNBF自身的水解反應)理論量。
2.2.3 衍生化反應時間的選擇
在保持衍生反應緩沖液pH值和衍生劑用量不變的情況下,于65 ℃水浴中分別衍生反應不同時間(30~60 min),觀察反應時間對色譜峰面積的影響。結果表明,隨著反應時間的延長,色譜峰面積呈上升趨勢,反應40 min后,硒代氨基酸的色譜峰面積已趨于最大。故本法選用40 min作為反應時間。
2.2.4 衍生反應緩沖液pH值的選擇
選用硼砂-硼酸緩沖液,比較不同pH值條件下(pH 8.6~11.0)進行衍生反應對硒代氨基酸峰面積的影響。結果表明,在pH 9.0時硒代氨基酸的峰面積最大。故將衍生介質的pH值定為9.0。
2.3 色譜條件的優化
SeMet和SeCys2與CNBF發生衍生化反應,衍生物在240 nm處產生紫外吸收。考慮到富硒米曲霉中含有其他氨基酸對硒代氨基酸的測定產生一定的干擾,采用乙腈-水(1∶1,V/V)和0.05 mol/L,pH 6.8乙酸-乙酸鈉緩沖液為流動相,根據米曲霉的特性對梯度洗脫的方法進行優化,最終在1.3.4節色譜條件下,使富硒米曲霉中的硒代氨基酸得到良好的分離。圖2為SeMet和SeCys2標準品的衍生色譜圖。
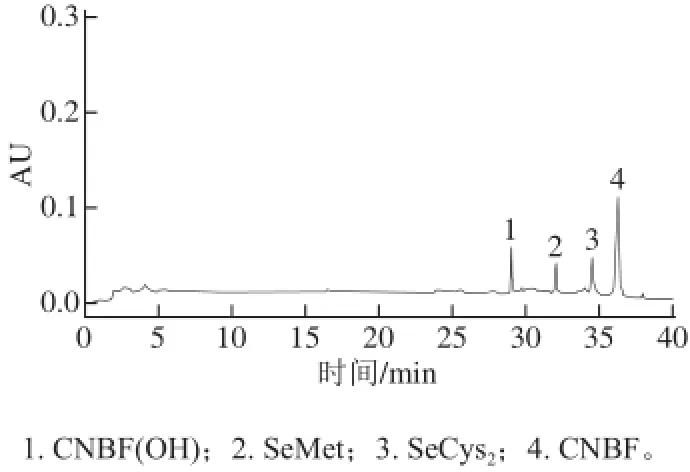
圖2 SeMet與SeCys衍生物的液相色譜圖Fig.2 Liquid chromatogram of the CNBF-SeMet and SeCys2derivatives
2.4 富硒米曲霉樣品的衍生色譜分析
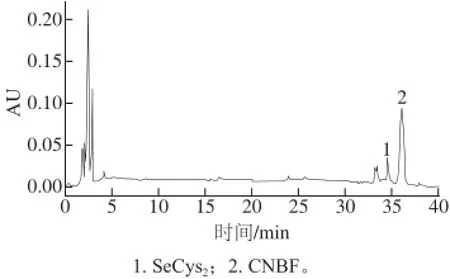
圖3 富硒米曲霉水解色譜圖Fig.3 Chromatogram of hydrolysate of selenium-enriched Aspergillus oryzae
富硒米曲霉在6 mol/L HCl溶液,50 ℃水解48 h后得到的產物進行衍生反應后的色譜分析如圖3所示。SeCys2得到了有效分離,但SeMet未出現明顯的色譜峰,可能是由于富硒米曲霉中含量過低或者是此提取方法對于SeMet的提取具有一定的局限性,具體原理有待進一步研究。圖中其余未標色譜峰可能是其他物質,由于缺少明確的標準物質,暫時無法確定。
2.5 方法的線性范圍與方法檢測限
取硒代氨基酸標準品儲備液逐級稀釋成5.0、10.0、20.0、40.0、80.0、100 mg/L的系列標準工作液,按上述試驗方法衍生后上機測定,以硒代氨基酸濃度為橫坐標,吸收峰面積為縱坐標做標準曲線,其檢測限及線性范圍見表2。

表2 硒代氨基酸的檢測限及線性范圍Table2 Linear calibration ranges, regression equations, R2, LOD anndd LOQ of CNFB-selenoamino acids derivatives
由表2可知,SeMet標準曲線y=12 059x-63.538,R2=0.997 5,測定質量濃度在5.0~100 mg/L內線性關系良好;SeCys2標準曲線y=17 432x-87.428,R2=0.992 8,測定質量濃度在5.0~100 mg/L也具有良好的線性關系,均可用于樣品的檢測分析。SeMet最低檢測限為0.41 mg/L,定量限為1.35 mg/L,其檢測靈敏度高于SeCys2的0.680 mg/L,高出39.7%,定量限也高于SeCys2的2.244 mg/L,高出66.2%。
2.6 方法的重復性及加標回收率實驗
取同一份富硒米曲霉樣品5 份,按照上述方法測定硒代氨基酸含量,5 次結果的相對標準偏差均小于5%,表明該方法重復性良好。
取富硒米曲霉樣品4 份,加入不同量SeCys2標準液,按照實驗方法測定,計算加標回收率。由表3可見,SeCys2有較高的加標回收率,為94.8%~105.8%,使得有機硒的測定真實可信,滿足富硒米曲霉測定的需要。

表3 富硒米曲霉中SeCCyyss2的加標回收率Table3 Spiked Recovery of SeCys2in selenium-enriched Aspergillus oryyzzaaee
3 結 論
實驗研究確立CNBF柱前衍生-反相高效液相色譜法檢測富硒米曲霉中有機硒形態的分析方法。重點對樣品的提取方法和衍生方法進行了研究。1)樣品的提取方法研究:比較不同溫度、不同時間對提取富硒米曲霉中硒代氨基酸的影響,并綜合考慮實驗條件的選取與實驗時間的消耗,采用一種溫和的實驗條件-6 mol/L HCl溶液,50 ℃水解48 h的方法,提取樣品中的硒化合物,在確保實驗定性定量結果的同時,提高了富硒米曲霉中硒代氨基酸的提取效率。2)硒代氨基酸衍生及分離研究:選擇CNBF柱前衍生,并對衍生劑用量、衍生時間以及衍生反應緩沖液的pH值進行了優化,采用乙腈-水(1∶1,V/V)和pH 6.8乙酸-乙酸鈉緩沖液為流動相進行梯度洗脫,使SeMet和SeCys2得到有效分離。該實驗方法的檢測限低、精密度高,線性相關性良好,加標回收率在94.8%~105.8%之間。與現在較為先進的HPLC-MS聯用分析硒代氨基酸的方法相比,具有普及性強,樣品處理簡單,對實驗設備要求低,同時兼顧準確性與經濟性的優勢,基本可以滿足硒形態分析的要求,能夠用于硒形態分析研究。
但同時實驗尚有不足,有待改進的地方。從色譜圖可以看出,目標物的出峰時間太晚,耽誤檢測效率,是否可以通過某些手段,使目標物的出峰時間提前,縮短檢測時間,提高實驗效率等,本課題組還將會對此領域的研究做進一步的探索。
[1] SCHRAUZER G N, WHITE D A. Elemental seleniumin organic selenium compounds: their chemistry and biology[J]. Bioinorganic Chemistry and Applications, 1983, 8(3): 303.
[2] WESTERMARK T. Selenium and selenides[J]. Acta Pharmacal Toxical, 1987, 4(1): 121.
[3] 潘艷. 富硒米曲霉發酵條件的探索[D]. 長春: 吉林大學, 2009.
[4] SMITH A M, PICCIANO M F. Relative bioavailability of selenocompounds in the laboratory rat[J]. Journal of Nutrition, 1987, 117(4): 725-731.
[5] 高愈希, 蒲云霞, 彭曉敏, 等. 反相離子對高效液相色譜-電感耦合等離子體質譜法測定富硒酵母中硒[J]. 理化檢驗: 化學分冊, 2013(6): 701-704.
[6] 程建中, 楊萍, 桂仁意. 植物硒形態分析的研究綜述[J]. 浙江農林大學學報, 2012(2): 288-295.
[7] 葉韻青. 富硒酵母類保健食品中有機硒測定方法[J]. 輕工科技, 2013(4): 7-8.
[8] 陳尚衛, 戴軍, 吳勝芳, 等. 富硒酵母中硒蛋氨酸的GC-MS/MS測定[J].食品與機械, 2012, 28(2): 60-63.
[9] 劉揚, 張濤, 劉靜, 等. 運用RPLC-ΙCP-MS對富硒酵母中硒形態分析[J].食品與生物技術學報, 2013, 32(12): 1261-1265.
[10] DENG Biyang, SHΙ Aihong, LΙ Linqiu, et al. Determination of selenomethionine in selenium-enriched yeast using capillary electrophoresis on-line coupled with electrochemiluminescence detection[J]. Microchim Acta, 2009, 165(3/4): 279-283.
[11] 仲娜. 電感藕合等離子體質譜(ΙCP-MS)及高效液相色譜與電感藕合等離子體質譜聯用技術用于富硒生物樣品中硒的化學形態組成及分布規律研究[D]. 青島: 中國海洋大學, 2007.
[12] SHΙ Tianyu, TANG Tao, QΙAN Kun, et al. High-performance liquid chromatographic method for determination of amino acids by precolumn derivatization with 4-chloro-3,5-dinitrobenzotrifl uoride[J]. Analytica Chimica Acta, 2009, 654: 154-161.
[13] 李玉玲, 李衛華. 反相高效液相色譜法檢測奶粉中含硫氨基酸[J].食品科學, 2012, 33(8): 167-170.
[14] 郝素娥, 滕冰. 硒酵母中有機硒及硒代氨基酸含量的測定方法[J].分析測試學報, 1999, 18(3): 72-74.
[15] 唐瑩瑩, 袁建, 蔣旭玲, 等. 柱前衍生-反相高效液相色譜法研究稻谷中有機硒形態[J]. 糧食與油脂, 2013, 26(12): 26-28.
[16] PEREIRA V, PONTES M, CAMARA J S, et al. Simultaneous analysis of free amino acids and biogenic amines in honey and wine samples using in loop orthophthalaldeyde derivatization procedure[J]. Journal of Chromatography A, 2008, 1189(1/2): 435-443.
[17] RIGOBELLO-MASINI M, PENTEADO J C P, LIRIA C W, et al. Implementing stepwise solvent elution in sequential injection chromatography for fluorimetric determination of intracellular free amino acids in the microalgae tetraselmis gracilis[J]. Analytica Chimica Acta, 2008, 628(2): 123-132.
[18] FERNANDEZ-FIGARES I, RODRIGUEZ L C, GONZALEZCASADO A. Effect of different matrices on physiological amino acids analysis by liquid chromatography: evaluation and correction of the matrix effect[J]. Journal of Chromatography B, 2004, 799(1): 73-79.
[19] 李東, 孫家義. 2,4-二硝基氟苯柱前衍生高效液相色譜法測定18 種氨基酸[J]. 化學分析計量, 2004, 13(1): 18-20.
[20] NAVAL M V, GOMEZ-SERRANILLOS M P, CARRETERO M E, et al. Value of high-performance liquid chromatographic analysis of amino acids in thedetermination of Panax ginseng radix ertract effect incultured neurons[J]. Journal of Chromatography A, 2006, 1121(2): 242-247.
[21] BOSCH L, ALEGRIA A, FARRE R. Application of the 6-aminoquinolyl-N-hydroxysccinimidyl carbamate (AQC) reagent to the RP-HPLC determination of amino acids in infant foods[J]. Journal of Chromatography B, 2006, 831(1/2): 176-183.
[22] KABELOVA I, DVORAKOVA M, CIZKOVA H, et al. Determination of free amino acids in beers:a comparison of Czech and foreign brands[J]. Journal of Food Composition and Analysis, 2008, 21(8): 736-741.
[23] LOPEZ-CERVANTES J, SANCHEZ-MACHADO D I, ROSASRODRIGUEZ J A. Analysis of free amino acids in fermented shrimp waste by high-performance liquid chromatography[J]. Journal of Chromatography A, 2006, 1105(1/2): 106-110.
[24] LOZNOV V, BENKOVA B, MATEVA L, et al. Liquid chromatography method for simultaneous analysis of amino acids and biogenic amines in biological fl uids with simultaneous gradient of pH and acetonitrile[J]. Journal of Chromatography B, 2007, 860(1): 92-97.
[25] 朱曙東, 趙昇皓. 氨基酸的高效液相色譜分析[J]. 色譜, 1994, 12(1): 20-24.
[26] FAN Xinjun, YOU Jinmao, KANG Jingwu, et al. New reagents for determination of amino acids by liquid chromatography with precolumn fl uorescence derivatization[J]. Analytica Chimica Acta, 1998, 367(1/2/3): 81-91.
[27] TODOROKI K, ETOH H, YOSHIDA H, et al. A fluorous tagbound fl uorescence derivatization reagent, F-trap pyrene, for reagent peak-free HPLC analysis of aliphatic amines[J]. Analytical and Bioanalytical Chemistry, 2009, 394(1): 321-327.
Determination of Organic Selenium Compounds in Selenium-Enriched Aspergillus oryzae by Pre-Column Derivatization-RP-HPLC Method
LI Hongwei1, WANG Kaiping1, WU Yu1, TANG Zhengjiang1, TANG Fei1, LIU Guoqing1,2,*
(1. School of Biotechnology and Food Engineering, Hefei University of Technology, Hefei 230009, China; 2. Anhui Wan Jiang Poultry Industry Research Institute, Xuancheng 242000, China)
A reversed phase-high performance liquid chromatography (RP-HPLC) method for the determination of organic selenium compounds in selenium-enriched Aspergillus oryzae was established. The extraction of organic selenium compounds from samples was performed through hydrolysis with 6 mol/L HCl solution for 48 h at 50 ℃. After pre-column derivatization with 4-chlorine-3,5-dinitrobenzotrifl uoride (CNBF), the derivatives were separated on a C18reversed-phase column through gradient elution with acetonitrile-water and pH 6.8 acetic acid-sodium acetate buffer solution. UV detection was carried out at 240 nm. The organic selenium compound in selenium-enriched A. oryzae was mainly selenocystine (SeCys2). The limit of detection (LOD) (RSN= 3) was 0.680 mg/L and the limit of quantitation (LOQ) (RSN= 10) was 2.244 mg/L. The linear range was 5.0-100 mg/L. The recoveries were in the range of 94.8%-105.8% and selenium-enriched A. oryzae contained 1.25 mg/g selenocystine. This method is simple, sensitive and accurate, and could be used to determine organic selenium compounds in selenium-enriched A. oryzae and related products.
selenium-enriched Aspergillus oryzae; organic selenium; selenoamino acids; pre-column derivatization-RP-HPLC
Q517
A
1002-6630(2015)22-0137-05
10.7506/spkx1002-6630-201522025
2015-01-07
安徽省科技攻關項目(1401032006)
李紅衛(1988—),男,碩士研究生,研究方向為食品質量與安全。E-mail:li3017545@163.com
*通信作者:劉國慶(1963—),男,教授,博士,研究方向為生物技術。E-mail:liugq_168@163.com
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
