UPLC—MS/MS建立雙黃連口服液中連翹苷、牛蒡子苷、灰氈毛忍冬皂苷乙、川續斷皂苷乙含量測定方法
2016-05-14 10:32:59顧珮馨殷愛玲
云南中醫中藥雜志 2016年5期
顧珮馨 殷愛玲
摘要:目的建立同時測定雙黃連口服液中連翹苷、牛蒡子苷、灰氈毛忍冬皂苷乙、川續斷皂苷乙4種有效成分含量的超高效液相色譜-串聯質譜(UPLC-MS/MS)分析方法。方法采用UPLC-MS/MS技術結合BEHC18(100 mm×2.1 mm,1.7 μm)色譜柱建立分析方法。利用乙腈-0.1%甲酸水梯度洗脫作為流動相,流速為0.4 mL·min-1,采用電噴霧電離源(ESI),多反應離子監測(MRM)掃描方式進行檢測。結果4種被測物呈良好的線性關系(r>0.999);精密度、重復性和穩定性良好;加樣回收率在99.12%~101.40%之間,RSD值均小于3%。結論 所建立的方法準確、快捷,重現性好,可用于雙黃連口服液中連翹苷、牛蒡子苷、灰氈毛忍冬皂苷乙、川續斷皂苷乙4種有效成分的含量的同時測定。
關鍵詞:超高效液相色譜-串聯質譜;雙黃連口服液;木脂素;皂苷類
中圖分類號:R284.2文獻標志碼:A文章編號:1007-2349(2016)05-0056-03
雙黃連口服液是由黃芩、金銀花、連翹通過一定傳統水提醇沉工藝精制而成,臨床主要用于細菌或病毒引起的上呼吸道感染。前期研究表明金銀花、連翹為方解雙黃連君藥,其抗菌、抗病毒效果顯著[1-2]。連翹苷、牛蒡子苷為連翹有效微量成分[3]。而灰氈毛忍冬皂苷乙、川續斷皂苷乙為金銀花有效微量成分[4-5]。由于HPLC-UV-ELSD分析條件的限制,使雙黃連口服液中這些微量成分無法追蹤。近些年,隨著質譜(MS)技術的發展。LC-MS技術用于中藥質量控制越來越多。本研究采用LC-MS/MS聯用技術,快速測定了雙黃連口服液中4種有效微量成分的含量,該方法具有準確、快捷、精密度高、靈敏性好等特點,為雙黃連制劑提供了一種簡單易行的定量方法,可用于該制劑的質量控制和臨床研究。
1儀器與試劑
1.1儀器設備超高效液相色譜-TQD質譜聯用儀(ACQUITY系統,美國Waters公司),MassLynx V4.1工作站。BP211D型電子分析天平(德國Sartorius公司),MicroCL 21R微量離心機(美國賽默飛世爾科技公司),微量移液器(上海科華實驗系統有限公司),默克密理博Synergy超純水儀(德國Merck公司)。
1.2藥品與試劑對照品牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙、川續斷皂苷乙(成都瑞芬思生物科技有限公司)均為供含量測定用,雙黃連口服液為哈藥制藥三廠生產,批號分別為12022231,10080188,13021633。甲醇(色譜純,德國Merck公司)、乙腈(色譜純,德國Merck公司),其余溶劑均為分析純,水為超純水。
2分析方法
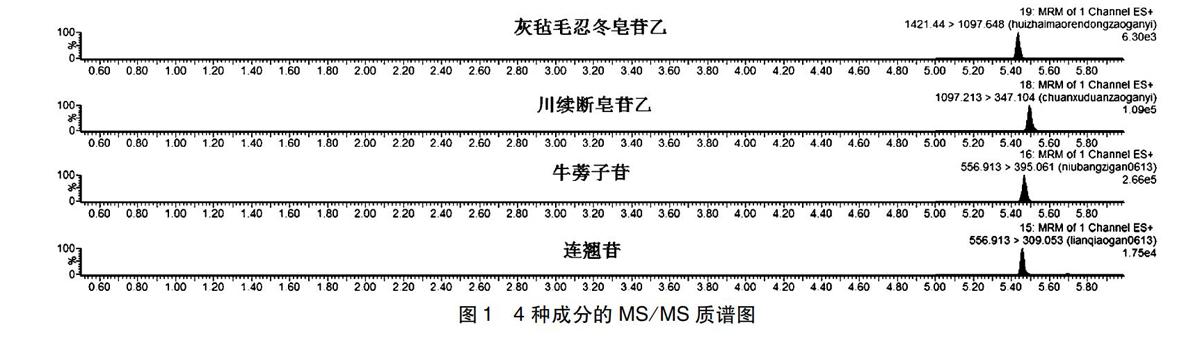
2.1色譜條件色譜柱:Waters Acquity BEH C18柱(100 mm×2.1 mm,1.7 μm)柱;流動相:A為乙腈,B為0.1%的甲酸水;梯度洗脫:0~1.5 min(90%B(90%B),1.5~4.0 min(90%B(40%B),4.0~4.2 min(40%B(10%B),4.2~5.2 min(10%B(10%B),5.2~7 min(10%B(90%B),7.0~8.0 min(90%B(90%B);流速:0.4 mL·min-1;進樣量:5 μL,柱溫40 ℃。見圖1。
2.2質譜條件Waters三重四級桿串聯質譜儀(TQD),離子化方式:電噴霧離子化(ESI),多反應監測離子掃描模式(MRM)測定;主要質譜參數為:毛細管電壓4KV,錐孔電壓60V,脫溶劑溫度450℃,脫溶劑氣體流速800L·Hr-1。四種成分離子對的選擇(M/Z):連翹苷:(ESI+,556.91>309.05),牛蒡子苷:(ESI+,556.91>395.06),灰氈毛忍冬皂苷乙:(ESI+,1421.44>1097.64),川續斷皂苷乙:(ESI+,[FL)]
2.3溶液的配制
2.3.1供試品溶液的制備取雙黃連口服液1 mL,置50 mL量瓶中,加50%甲醇20 mL,超聲(35kHz)處理20分鐘,放置至室溫,加50%甲醇稀釋至刻度,搖勻。12,000 r/min離心10 min,取上清。用10%(乙腈:甲醇(4:1))含0.4%甲酸及0.5 mM甲酸鈉溶液稀釋50倍,過0.22 μm有機膜,取上清液5 μL,UPLC-ESI-MS/MS分析。
2.3.2混合對照品溶液的制備精密稱取牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙和川續斷皂苷乙對照品適量,用甲醇(0.1%甲酸)制成每1 mL含牛蒡子苷1.10 μg,連翹苷2.29 μg,灰氈毛忍冬皂苷乙4.81 μg,川續斷皂苷乙3.89 μg。4℃保存,備用。
3方法學考察
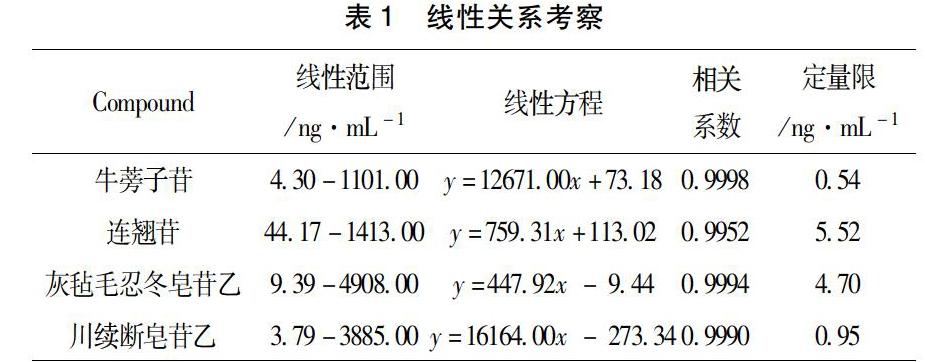
3.1線性關系與定量限分別精密量取“2.4.2”項下的混合對照品溶液適量,置于10 mL容量瓶中,用初始流動相乙腈-0.1%甲酸水(1:9,v/v)稀釋成系列濃度的混合對照品溶液(牛蒡子苷:1100,550,275,137.5,68.75,34.38,17.19,8.59,4.30,2.15 ng·mL-1;連翹苷:2290,1145,572.5,286.25,143.13,71.56,35.78,17.89,8.95,4.47 ng·mL-1;灰氈毛忍冬皂苷乙:4810,2405,1202.5,601.25,300.63,150.31,75.16,37.58,18.79,9.39 ng·mL-1;川續斷皂苷乙:3890,1945,972.5,486.25,243.13,121.56,60.78,30.39,15.20,7.60 ng·mL-1)。按照“2.1和2.2”項下的條件進樣5 μL測定。以被測組分的質量濃度X(ng·ml-1)為橫坐標,以各成分的分子離子峰峰面積均值 Y 為縱坐標進行線性回歸,得到各成分的回歸方程、相關系數及線性范圍,以信噪比(S/N)為10來確定各待測組分的定量限。表1顯示牛蒡子苷在4.30-1101.00 ng·mL-1范圍內相關系數為0.9998,連翹苷在44.17-1413.00 ng·mL-1范圍內相關系數為0.9952,灰氈毛忍冬皂苷乙在9.39-4908.00 ng·mL-1范圍內相關系數為0.9994,川續斷皂苷乙在3.79-3885.00 ng·mL-1范圍內相關系數為0.9990。且定量限分別為0.54,5.52,4.70,0.95 ng·mL-1。以上結果表明各成分在各自的線性范圍內具有良好的線性關系,且定量限較低。
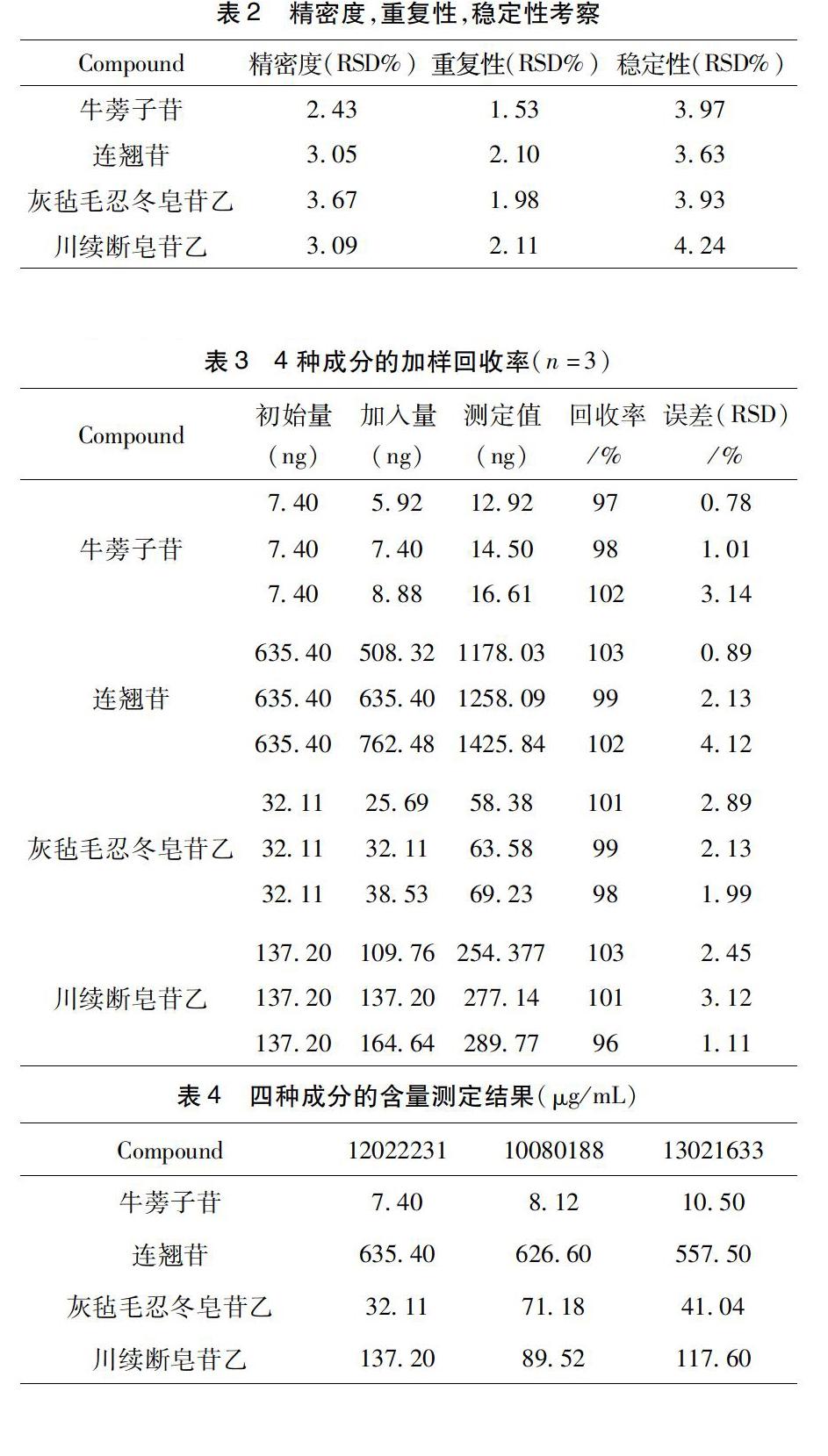
3.2精密度考察取供試品溶液,重復進樣6次,依法測定。結果顯示,牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙和川續斷皂苷乙的峰面積相對標準偏差(RSD)值分別為2.43,3.05,3.67,3.09%。結果(表2)表明該方法的精密度良好。
3.3重復性考察按“2.4.1”項下方法制備供試品(12022231)6份,,按“2.1和2.2”條件進樣分析,測得此批號中牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙和川續斷皂苷乙平均含量分別為7.40,635.40,32.11,137.20 μg/mL。RSD值分別為1.53,2.10,1.98,2.11%,結果(表2)表明該方法的重復性良好。
3.4穩定性實驗取供試品溶液,分別于0,2,4,6,8,10,12,24 h進樣測定,測得牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙和川續斷皂苷乙峰面積的RSD值分別為3.97,3.63,3.93,4.24%。結果(表2)表明供試品溶液在24h內穩定。
3.5加樣回收率實驗精密吸取已知濃度的供試品溶液9等份,每3份為一組,分別精密加入高、中、低3個濃度的混合標準品溶液后,按“2.4.1”項下方法制備供試品溶液進行測定。計算各成分的回收率。本方法在加樣3個濃度水平下,四種成分的加樣回收率在95.00%~105%之間,RSD值均小于3%,能夠滿足雙黃連口服液中四種有效成分的含量測定要求。結果見表3。
3.6樣品含量測定取3個批號的雙黃連口服液,按照“2.3.1”項下方法處理,平行制得各組待測樣品溶液3份,進樣分析,經計算得牛蒡子苷,連翹苷,灰氈毛忍冬皂苷乙,川續斷皂苷乙的含量。具體見表4。
[HT64結論
4.1雙黃連口服液的化學成分研究甚多,但多集中于運用HPLC[6]技術來檢測并追蹤化學成分,并對制劑中具較強紫外吸收且含量較高的成分進行定量分析,但對弱紫外吸收,含量較低且較難分離的成分含量測定分析時則相對困難。作者經過查閱文獻發現,目前尚未見到報道用液-質串聯的方法同時檢測雙黃連口服液中多種微量成分含量的報道,本實驗首次使用LC-MS/MS聯用技術,對雙黃連口服液中多種微量成分進行了同時測定。方法簡單、可靠、應用性強。
4.2本實驗同時選用正離子模式測定4種成分的含量,且分子離子峰均為[M+Na]+。實驗過程發現這四種物質[M+Na]+豐度強且穩定。MS/MS亦發現穩定且靈敏度高。
4.3本實驗建立的液-質聯用方法,可在短時間內快速分離檢測雙黃連口服液中牛蒡子苷、連翹苷、灰氈毛忍冬皂苷乙和川續斷皂苷乙4種微量成分,該方法經考察,準確,快捷,重現性好,符合中藥制劑的相關分析要求,可用于雙黃連口服液的質量控制。此外,雙黃連口服液的體內研究相對較少,隨著液-質聯用技術的不斷普及,雙黃連口服液物質基礎研究包括體內研究也將越來越多。本實驗為雙黃連口服液的質量控制提供了依據,且為該制劑的臨床前與臨床藥代動力學等相關研究提供了一定的參考。
4.4中藥制劑定性、定量研究最初為單一指標性成分,隨著以色譜質譜聯用為代表(LC-MS/MS)的各種分析儀器、分離手段和計算機技術的迅速發展,目前,中藥制劑化學成分定[LL]性、定量研究已經廣泛采用多指標成分,為闡明中藥的物質基礎提供了重要手段[7]。由此可見,不斷應用新理論、新方法和新技術將會有力的推動中藥的現代研究,大大加快對中藥物質基礎與質量控制的研究。[KH*1D]
參考文獻:
[1]Shang XF.,Pan H.,Li MX.,et al.Lonicera japonica Thunb.:Ethnopharmacology,phytochemistry and pharmacology of an important traditional Chinese medicine.J.Ethnopharmacol.2011,138:1-21.
[2]Zhou,W.,Tan,X.B.,Shan,J.J.,et al.Study on the Main Components Interaction from Flos Lonicerae and Fructus Forsythiae and Their Dissolution In Vitro and Intestinal Absorption in Rats.2014,Plos one.9,e109619.
[3]方穎,鄒國安,劉焱文.連翹的化學成分[J].中國天然藥物,2008,6(3):235-236.
[4]婁紅祥,郎偉君,呂木堅.金銀花中水溶性化合物的分離與結構確定[J].中草藥,1996,27(4):195.
[5]陳昌祥,王薇薇,倪偉,等.金銀花花蕾中的新三帖皂苷[J].云南植物研究,2000,22(2):201-208.
[6]陳國寶,宋桂萍,楊棄塵.HPLC法同時測定雙黃連片中黃芩苷、綠原酸、連翹苷、連翹酯苷的含量[J].中國藥房,2011,48:4594-4596.
[7]Wang XJ.,Zhang AH.,Yan GL.,et al.UHPLC-MS for the analytical characterization of traditional Chinese medicines[J].Trends Analyt.Chem.2014,63:180-187.
