一患青年型糖原貯積癥Ⅱ型姐弟的家系GAA基因新突變分析*
2016-07-15 08:40:53徐玲玲梁玉堅黃雪瓊張麗丹裴瑜馨程玉才
重慶醫學 2016年18期
關鍵詞:基因突變
徐玲玲,唐 雯△,梁玉堅,張 成,黃雪瓊,張麗丹,裴瑜馨,程玉才
(中山大學附屬第一醫院:1.PICU;2.神經內科,廣州 510080)
?
一患青年型糖原貯積癥Ⅱ型姐弟的家系GAA基因新突變分析*
徐玲玲1,唐雯1△,梁玉堅1,張成2,黃雪瓊1,張麗丹1,裴瑜馨1,程玉才1
(中山大學附屬第一醫院:1.PICU;2.神經內科,廣州 510080)
[摘要]目的鑒定一患青年型糖原貯積癥Ⅱ型(GSD Ⅱ)姐弟的家系酸性α-葡萄糖苷酶(GAA)基因的新致病性突變。方法對因“反復呼吸道感染、呼吸衰竭伴全身肌無力”就診的一姐弟倆的臨床及家系資料進行分析,均經α-1,4 GAA活性測定確診為青年型GSD Ⅱ,并提取先證者及其父母的外周血脫氧核糖核酸(DNA),聚合酶鏈反應(PCR)擴增GAA基因的全部20個外顯子及剪接位點序列,對PCR產物進行直接測序。結果弟弟GAA基因有2個復合雜合性突變,為遺傳自父親的外顯子8的c.1216G>A(p.Asp406Asn)錯義突變和遺傳自母親的外顯子14的c.1935C>A(p.Asp645Glu)錯義突變。結論GAA基因的c.1216G>A和c.1935C>A復合雜合性突變導致了該患兒出現以呼吸困難及心肌肥厚為特征的青年型GSD Ⅱ,新的突變c.1216G>A突變可能與青年型GSD Ⅱ相關。
[關鍵詞]糖原貯積癥Ⅱ型;pompe病;青年型;酸性α-葡萄糖苷酶;新突變;c.1216G>A;c.1935C>A
糖原貯積癥Ⅱ型(glycogen storage disease Ⅱ,GSD Ⅱ)又稱Pompe病,是一種由于酸性α-葡萄糖苷酶(acid alpha-glucosidase,GAA)或酸性麥芽糖酶(EC 3.2.1.20,Swiss-Prot P10253)缺陷引起的常染色體隱性溶酶體儲積癥。根據發病年齡,GSDⅡ可分為嬰兒型和晚發型;其中嬰兒型又可分為經典及非經典兩類,晚發型可進一步分為青少年及成年起病型[1]。嬰兒型癥狀最為嚴重,快速進展的骨骼肌無力所致呼吸衰竭及心肌肥大所致心力衰竭為其特征性表現;如不及時治療,多在1歲內死亡。青年型患者表現為緩慢進展的肌無力或進行性肌營養不良,常累及呼吸肌需要機械通氣,極少累及心肌[2]。GAA酶活性下降程度與發病年齡及GSDⅡ疾病的嚴重程度有關[3]。GAA 酶活性降低是由GAA 基因(NM_000152)突變引起的。GSDⅡ的確診依賴于GAA酶活性的測定或GAA 基因分析。GSDⅡ普遍發病率大約為1∶40 000[4]。中國臺灣GSDⅡ新生兒的發病率為1∶20 000~40 000[5]。中國臺灣嬰兒型GSDⅡ的發病率大約為1∶33 333[6]。迄今中國大陸僅有少數幾篇關于GAA 突變分析的報道[7-14]。本文對一患有青年型GSDⅡ的姐弟倆的家系GAA基因的新致病性突變(c.1216G>A,c.1935C>A)進行分析。
1資料與方法
1.1一般資料本項目研究對象為臨床診斷為 GSDⅡ的一家系(二代人,共5人)一患病姐弟及健康的父母和姐姐(圖1)。該家系來自福建三明市,漢族。先證者父母為非近親婚配,其母孕期體健,先證者父母和姐姐有均無心臟、骨骼或肌肉的異常表現,血緣親屬中無相應病史。先證者1:男,弟弟,2歲,2012 年12月出生,足月順產,出生體質量4.7 kg,出生時無窒息搶救史,因“咳嗽、呼吸困難并全身肌肉無力2個月余”于2014 年12 月27日入住中山大學附屬第一醫院兒童重癥監護病房搶救。患兒3個月抬頭,6個月坐,1歲能扶站,1歲8個月能獨立行走。入院查體: 機控呼吸20次/分,體質量9.0 kg(-3.0SD),身高95 cm(0.2SD),消瘦,身高別體質量-3.2SD[15],未見明顯的肝脾腫大及舌體肥大。神經系統檢查雙上肢肌力2或3級,雙下肢肌力2級,膝腱及跟腱反射消失。血肌酸激酶升高為201~1 766 U/L(正常值25~200 U/L)。心臟彩超提示肥厚性心肌病。肌電圖顯示左側股直肌、左側肱二頭肌、雙側脛骨前肌及右側腓腸肌神經源性損傷。先證者2:女,姐姐,4歲,2010 年12月出生,足月順產,出生體質量3.9 kg,出生時無窒息搶救史,因“反復呼吸道感染、呼吸困難并全身肌肉無力1個月余”于2014 年12 月30日入住中山大學附屬第一醫院兒童重癥監護病房搶救。患兒3個月抬頭,6個月坐,1歲能扶站,1歲2個月能獨立行走。入院查體:機控呼吸20次/分,體質量12.0 kg(-3.5SD),身高95 cm(-2.0SD),消瘦,身高別體質量-2.2SD[15],未見明顯的肝脾腫大及舌體肥大。神經系統檢查雙上肢肌力4級,雙下肢肌力3級,膝腱及跟腱反射減弱。血肌酸激酶升高(237~803 U/L)。心臟彩超提示左心室肥大。肌電圖顯示左側腓腸肌、雙側肱二頭肌及脛骨前肌神經源性損傷。
1.2方法
1.2.1GAA酶活性測定用外周血白細胞法檢測[16]。
1.2.2提取DNA基因組DNA的提取經患者家屬知情同意,并經中山大學倫理委員會批準后,取先證者1及其父母外周血2 mL,應用過柱法(QIAamp Blood DNA Mini Kit, QIAGEN公司,美國)提取外周血DNA。
1.2.3對患者及其父母和姐姐的GAA基因序列分析基因的全部20個外顯子及剪接位點進行直接序列分析。應用Primer Premier 5.0軟件針對GAA基因的外顯子編碼區設計引物,應用2 X PCR MasterMix聚合酶(TIANGEN,天根)進行PCR擴增(ABI9700型PCR儀,Life technology,美國),然后對PCR產物進行直接測序(ABI3500測序儀,Life technology,美國),與參考序列(NG_009822.1與NM_000152.3)進行比較,從而發現可能存在的基因突變。變異位點的描述按人類基因組變異協會所推薦的命名法則(http://www.Hgvs.org/mutnomen/)[17]。
2結果
2.1先證者GAA酶活性水平先證者1的GAA酶活性為1.45 nmol·h-1·mg-1(正常值為大于14.00 nmol·h-1·mg-1),先證者2的GAA酶活性為1.03 nmol·h-1·mg-1,均診斷為青年型GSDⅡ。
2.2先證者1的GAA基因先證者1的GAA 基因有兩個復合雜合性突變:c.1216G>A 和c.1935C>A。其中位于第8外顯子的c.1216G>A突變,使第406位密碼子GAC變為AAC,導致正常的天門冬氨酸(Asp)變為天冬酰胺(Asn),即p.Asp406Asn錯義突變(圖2A);位于第14外顯子的c.1935C>A突變,使第645位密碼子GAC變為GAA,導致正常的天門冬氨酸(Asp)變為谷氨酸(Glu),即p.Asp645Glu錯義突變(圖2B)。其中c.1216G>A 突變來自父親,父親為該突變的雜合子;c.1935C>A 突變來自母親,母親為該突變的雜合子。先證者2及未患病的姐姐因其父母不同意,未行基因檢測。由于GAA 基因位于常染色體上,該突變在此家系中的傳遞符合孟德爾遺傳規律,遺傳方式符合常染色體隱性遺傳病的特征。GAA 基因的c.1216G>A 和c.1935C>A復合雜合性突變導致了姐弟倆出現以呼吸困難及心肌肥大為特征的青年型GSDⅡ。先證者父母因社會因素拒絕給先證者2及未患病姐姐行基因學檢查。兩位先證者住院期間予小潮氣量機械通氣,抗生素靜滴控制肺部感染,加強翻身拍背吸痰,營養神經、護心、護肝等對癥支持治療后,均很快撤離呼吸機,感染控制后出院。現姐姐可自行爬樓梯,弟弟可自行行走。兩者因為社會因素均未使用酶治療。
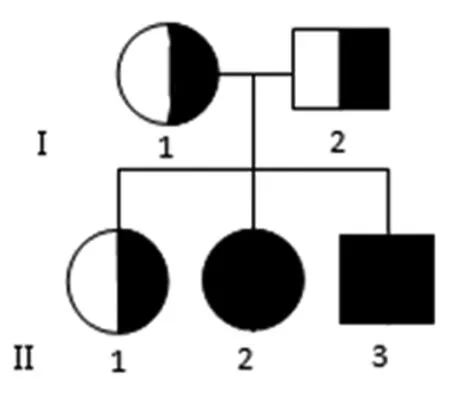
圖1 一個青年型GSDⅡ的家系
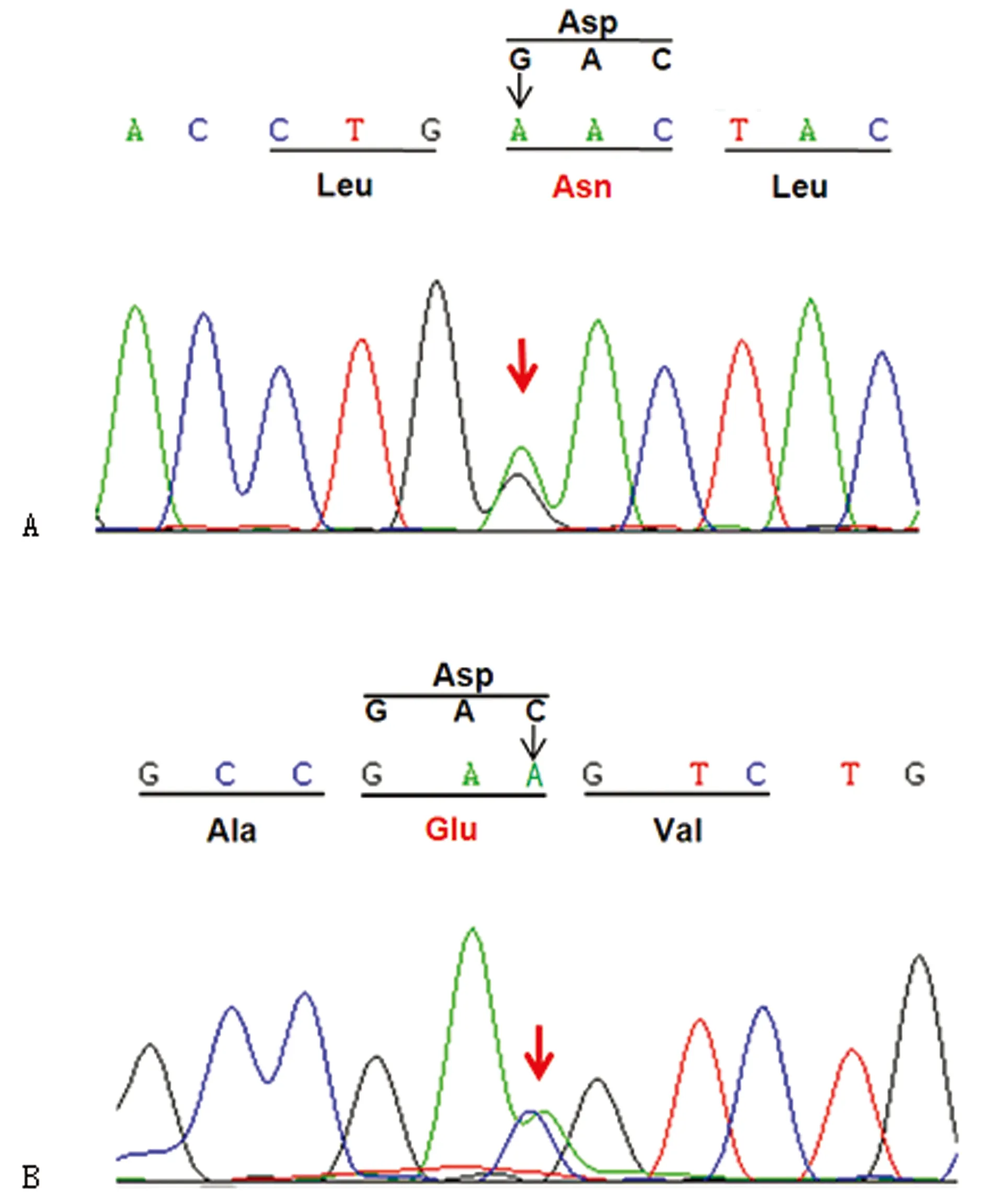
A:患者及其父親的 c.1216G>A(p.Asp406Asn)基因突變;B:患者及其母親的 c.1935C>A(p.Asp645Glu)基因突變。
3討論
GSDⅡ是由于α-1,4葡萄糖苷酶缺乏引起一種常染色體隱性遺傳病[18]。目前GSDⅡ在世界范圍內均有發病,約為1∶40 000~1∶146 000[4];中國大陸地區僅見散發病例報道。GAA基因是GSDⅡ的致病基因4,位于17q25.3,長約28 Kb,包含20個外顯子。GAA酶是由952個氨基酸組成(http://genome.ucsc.edu)的相對分子質量為105 000 U的酶蛋白,包含7個不同部位的樹狀結構,其前體需經過廣泛的翻譯后修飾,才成為成熟的酶[18]。
迄今全世界已發現的引起GSDⅡ的GAA基因突變500種(http://www.hgmd.cf.ac.uk,HGMD Professional,2014.09)。GAA基因突變主要集中在基因的3個臨界區域:包含起始密碼子的外顯子2,包含酶催化部位的外顯子10和11,及與蛋白高度保守區域相對應的14號外顯子[19]。突變類型主要為錯義突變及無義突變等[20]。GAA基因突變具有種族差異。如最常見的突變為高加索患者的c.-32-13T>G突變[21]。然而c.1935C>A(p.Asp645Glu)和c.2238G>C(Trp746Cys)是中國臺灣患者常見的突變類型[8,22],但這兩突變不存在日本人中[23]。在中國大陸GAA突變所致GSDⅡ僅見散發病例報道。Fu等[8]學者對18例中國嬰兒型GSDⅡ的患兒進行基因分析,發現中國大陸6種新突變(c.1356delC,c.378G>A,c.1827C>G,c.859-2 A>T,c.1551+2T>G,and c.1465G>T)。本研究報道中國大陸一青年型GSDⅡ家系的兩個新突變基因c.1935C>A(p.Asp645Glu)和c.1216G>A(p.Asp406Asn)。c.1216G>A突變尚少見在國內外患者中報道。c.1935C>A突變已被證實可引起突變等位基因編碼的蛋白殘存的GAA酶活性嚴重減少。
本研究報道的這一青年型GSDⅡ的姐弟除了有反復呼吸道感染、呼吸困難、呼吸肌及四肢肌肉無力主要表現這些癥狀外,同時合并心肌的受累。呼吸困難是青年型GSDⅡ常見的臨床表現,常常進展為呼吸衰竭,需要呼吸機輔助通氣治療[24]。有研究表明,晚發型(青年型和成年型)小于10%的患者出現心血管受累,如心電生理的異常和心肌肥大[25]。本研究患病姐姐存在左心室肥大;患病弟弟存在肥厚型心肌病。這在晚發型中是少見的。本研究在治療上予小潮氣量機械通氣、抗生素抗感染、加強翻身拍背吸痰,同時營養神經、護心、護肝等對癥支持治療,患兒很快撤離呼吸機。由于社會因素,兩例患兒均未予酶替代治療。
本研究發現,先證者(即弟弟)遺傳了母親的c.1935C>A(p.Asp645Glu)突變基因和父親的c.1216G>A(p.Asp406Asn)突變基因。其父母均為無癥狀的攜帶者。錯義突變c.1216G>A,位于第8外顯子,可能為不全或低外顯率突變基因,需注意基因的多態性,但結合臨床可以確診為GSDⅡ,且目前尚未被報道。另一個錯義突變c.1935C>A,位于外顯子14,已在臺灣患者中發現[26]。現在在中國大陸已有早發型和晚發型患者發現c.1935C>A突變基因文獻[8,27]。錯義突變被報道與酶的合成、轉運、翻譯后修飾及功能有關[28]。Shieh等[29]報道中國患兒80%存在Asp-645→Glu改變。基因突變導致Asp-645→Glu被證實影響合成α葡萄糖苷酶前體的轉運、磷酸化和蛋白酶加工的過程,最終導致α葡萄糖苷酶活性下降67%[30]。c.1216G>A基因突變導致患病的可能原因有幾種。(1)在多重序列比對分析中發現,在數種哺乳動物中該酶均存在氨基酸高度保守區域,該保守區域改變可能導致患病。(2)該突變導致帶電荷的天門冬氨酸變為不帶電荷的天冬酰胺,可能導致蛋白質構象的改變從而影響酶的活性。研究也發現許多錯義突變產生單一的氨基酸的改變在遲發型GSDⅡ患者中多見[31]。
本研究中,患兒父親攜帶c.1216G>A基因突變,母親攜帶c.1935C>A,均為雜合狀態,臨床上無癥狀。弟弟有c.1935C>A(p.Asp645Glu)和c.1216G>A(p.Asp406Asn)兩個基因突變。不幸的是,健康姐姐及患病姐姐的基因因社會因素未能檢測。結合患病的情況,推測患病姐姐攜帶同弟弟一樣的兩個基因突變。健康姐姐的基因突變可能與父親或母親的一樣。文獻報道GAA酶活性下降程度與發病年齡與GSDⅡ疾病的嚴重程度有關[32]。但臨床上發現弟弟較患病姐姐發病早,病情重,GAA酶活性較姐姐的高。已有的報道發現中國患者c.1935C>A基因突變所致的臨床表型存在變異性。而且文獻報道,GSDⅡ的臨床表型除了受基因型影響,還受其他因素,如生后的環境因素來影響疾病的臨床表現及疾病病程[33]。因此,c.1216G>A基因突變導致Pompe病的臨床表型難以確定。
Myozyme(重組GAA酶)替代治療是目前嬰兒型及晚發型GSDⅡ的惟一特異性治療方法;治療越早,療效越顯著[34-35]。但治療費用極其昂貴、部分患者存在免疫反應及神經肌肉的不可修復損害等。
總之,本研究通過酶學及基因學確診了中國大陸一患青年型GSD Ⅱ姐弟,發現一新的雜合GAA基因突變c.1216G>A和c.1935C>A。這對遲發型GSD Ⅱ來說,該基因突變的發現,對以后早期診斷具有重要意義。
參考文獻
[1]Tura?a LT,De Faria DO,Kyosen SO,et al.Novel GAA mutations in patients with Pompe disease[J].Gene,2015,561(1):124-131.
[2]Crescimanno G,Modica R,Lo Mauro R,et al.Role of the cardio-pulmonary exercise test and six-minute walking test in the evaluation of exercise performance in patients with late-onset Pompe disease[J].Neuromuscul Disord,2015,25(7):542-547.
[3]Todd AG,Mcelroy JA,Grange RW,et al.Correcting neuromuscular deficits with gene therapy in pompe disease[J].Ann Neurol,2015,78(2):222-234.
[4]Martiniuk F,Chen A,Mack A,et al.Carrier frequency for glycogen storage disease type Ⅱ in New York and estimates of affected individuals born with the disease[J].Am J Med Genet,1998,79(1):69-72.
[5]Lin CY,Shieh JJ.Molecular study on the infantile form of Pompe disease in Chinese in Taiwan[J].Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi,1996,37(2):115-121.
[6]Chien YH,Chiang SC,Zhang XK,et al.Early detection of Pompe disease by newborn screening is feasible:results from the Taiwan screening program[J].Pediatrics,2008,122(1):e39-45.
[7]Liu X,Wang Z,Jin W,et al.Clinical and GAA gene mutation analysis in mainland Chinese patients with late-onset Pompe disease:identifying c.2238G>C as the most common mutation[J].BMC Med Genet,2014,15(1):141.
[8]Fu L,Qiu W,Yu Y,et al.Clinical and molecular genetic study of infantile-onset Pompe disease in Chinese patients:identification of 6 novel mutations[J].Gene,2014,535(1):53-59.
[9]傅立軍,竇薇,周愛卿,等.糖原累積病Ⅱ型的臨床分析和基因學檢測[J].臨床兒科雜志,2006,24(12):962-965.
[10]操基清,張成,劉友章,等.糖原貯積癥Ⅱ型一家系的臨床特點及基因突變分析[J].中華神經科雜志,2013,46(1):32-36.
[11]陳素琴,陳路明,田秋紅,等.嬰兒肥厚型心肌病一家系GAA基因的突變分析[J].中山大學學報(醫學科學版),2014,35(1):139-143.
[12]仇佳晶,魏珉,張為民,等.幼年起病的2例晚發型糖原貯積癥Ⅱ型/Pompe病臨床和基因分析[J].中華兒科雜志,2007,45(10):760-764.
[13]曾敏慧,邱文娟,顧學范,等.一個糖原累積病Ⅱ型家系的酸性-α-葡萄糖苷酶及其產前基因診斷[J].中華醫學遺傳學雜志,2011,28(3):261-265.
[14]曾敏慧,邱文娟,顧學范,等.1例糖原累積病患兒酸性-α-葡萄糖苷酶基因的新無義突變p.W738X[J].臨床兒科雜志,2011,29(5):401-406.
[15]李輝,季成葉,宗心南等.中國0~18歲兒童、青少年身高、體重的標準化生長曲線[J].中華兒科雜志,2009,47(7):487-492.
[16]Jack RM,Gordon C,Scott CR,et al.The use of acarbose inhibition in the measurement of acid alpha-glucosidase activity in blood lymphocytes for the diagnosis of Pompe disease[J].Genet Med,2006,8(5):307-312.
[17]Den Dunnen JT,Antonarakis SE.Mutation nomenclature extensions and suggestions to describe complex mutations:a discussion[J].Hum Mutat,2000,15(1):7-12.
[18]Sugawara K,Saito S,Sekijima M,et al.Structural modeling of mutant alpha-glucosidases resulting in a processing/transport defect in Pompe disease[J].J Hum Genet,2009,54(6):324-330.
[19]Fernandez-Hojas R,Huie ML,Navarro C,et al.Identification of six novel mutations in the acid alpha-glucosidase gene in three Spanish patients with infantile onset glycogen storage disease type Ⅱ (Pompe disease)[J].Neuromuscul Disord,2002,12(2):159-166.
[20]Kroos M,Hoogeveen-Westerveld M,Van Der Ploeg A,et al.The genotype-phenotype correlation in Pompe disease[J].Am J Med Genet C Semin Med Genet,2012,160C(1):59-68.
[21]Kroos MA,Pomponio RJ,Hagemans ML,et al.Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype[J].Neurology,2007,68(2):110-115.
[22]Amarinthnukrowh P,Tongkobpetch S,Kongpatanayothin A,et al.p.D645E of acid α-glucosidase is the most common mutation in thai patients with infantile-onset pompe disease[J].Genet Test Mol Biomarkers,2010,14(6):835-837.
[23]Raben N,Plotz P,Byrne BJ.Acid alpha-glucosidase deficiency (glycogenosis type Ⅱ,Pompe disease)[J].Curr Mol Med,2002,2(2):145-166.
[24]Smith BK,Collins SW,Conlon TJ,et al.Phase Ⅰ/Ⅱ trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease:initial safety and ventilatory outcomes[J].Hum Gene Ther,2013,24(6):630-640.
[25]Van Der Beek NA,Soliman OI,Van Capelle CI,et al.Cardiac evaluation in children and adults with Pompe disease sharing the common c.-32-13T>G genotype rarely reveals abnormalities[J].J Neurol Sci,2008,275(1/2):46-50.
[26]Amarinthnukrowh P,Tongkobpetch S,Kongpatanayothin A,et al.p.D645E of acid α- glucosidase is the most common mutation in thai patients with infantile-onset pompe disease[J].Genet Test Mol Biomarkers,2010,14(6):835-837.
[27]Yang CC,Chien YH,Lee NC,et al.Rapid progressive course of later-onset Pompe disease in Chinese patients[J].Mol Genet Metab,2011,104(3):284-288.
[28]Hermans MM,De Graaff E,Kroos MA,et al.The effect of a single base pair deletion (delta T525) and a C1634T missense mutation (pro545leu) on the expression of lysosomal alpha-glucosidase in patients with glycogen storage disease type Ⅱ[J].Hum Mol Genet,1994,3(12):2213-2218.
[29]Shieh JJ,Wang LY,Lin CY.Point mutation in Pompe disease in Chinese[J].J Inherit Metab Dis,1994,17(1):145-148.
[30]Hermans MM,De Graaff E,Kroos MA,et al.The conservative substitution Asp-645——>Glu in lysosomal alpha-glucosidase affects transport and phosphorylation of the enzyme in an adult patient with glycogen-storage disease type Ⅱ[J].Biochem J,1993,289(Pt 3):687-693.
[31]Pittis MG,Montalvo AL,Miocic S,et al.Identification of four novel mutations in the alpha glucosidase gene in five Italian patients with infantile onset glycogen storage disease type Ⅱ[J].Am J Med Genet A,2003,121(3):225-230.
[32]Remiche G,Ronchi D,Magri F,et al.Extended phenotype description and new molecular findings in late onset glycogen storage disease type Ⅱ:a northern Italy population study and review of the literature[J].J Neurol,2014,261(1):83-97.
[33]Wens SC,van Gelder CM,Kruijshaar ME,et al.Phenotypical variation within 22 families with Pompe disease[J].Orphanet J Rare Dis,2013,8(1):182.
[34]Case LE,Beckemeyer AA,Kishnani PS.Infantile Pompe disease on ERT:update on clinical presentation,musculoskeletal management,and exercise considerations[J].Am J Med Genet C Semin Med Genet,2012,160(1):69-79.
[35]Chien YH,Hwu WL,Lee NC.Pompe disease:early diagnosis and early treatment make a difference[J].Pediatr Neonatol,2013,54(4):219-227.
Analysis on novel mutations in GAA gene of a Chinese family with two siblings affected with juvenile onset form glycogen storage disease Ⅱ*
XuLingling1,TangWen1△,LianYujiang1,ZhangCheng2,HuangXueqiong1,ZhangLidan1,PeiYuxin1,ChengYucai1
(1.PediatricIntensiveCareUnit;2.DepartmentofNeurology,FirstAffiliatedHospitalofSunYat-senUniversity,Guangzhou,Guangdong510080,China)
[Abstract]ObjectiveTo identify a novel pathogenicity mutation of acid alpha-glucosidase(GAA) gene in a Chinese family with two siblings affected with juvenile onset form glycogen storage disease Ⅱ(GSD Ⅱ).MethodsThe clinical and family data of two siblings presenting recurrent respiratory tract infections,respiratory failure associated with systemic muscle weakness,were analyzed and diagnosed with GSDⅡ by detecting alpha-1,4-glucosidase activity.DNA was extracted from peripheral blood of the proband,younger brother and his parents.All 20 exons and the intron-exon splice sites of GAA gene were amplified by polymerase chain reaction (PCR).Mutations were detected by direct sequencing the PCR products.ResultsThe younger brother was found to be compound heterozygous for two mutations in the GAA gene:c.1216G>A (p.Asp406Asn) missense mutation in the exon 8 from his father and c.1935C>A (p.Asp645Glu) missense mutation in the exon 14 from his mother.ConclusionThe compound heterozygous c.1216G>A and c.1935C>A mutations caused the juvenile onset form GSDⅡ characterized by dyspnea and cardiac hypertrophy.The novel c.1216G>A mutation may be related to the juvenile onset form GSDⅡ.
[Key words]glycogen storage disease Ⅱ;pompe disease;juvenile onset;acid alpha-glucosidase;novel mutation;c.1216G>A;c.1935C>A
doi:·論著·10.3969/j.issn.1671-8348.2016.18.004
基金項目:2010年國家臨床重點專科建設項目(部183)。
作者簡介:徐玲玲(1985-),主治醫師,碩士研究生,主要從事兒童內分泌及重癥醫學的研究。△通訊作者,E-mail:tangwen@mail.sysu.edu.cn。
[中圖分類號]R394.3
[文獻標識碼]A
[文章編號]1671-8348(2016)18-2460-04
(收稿日期:2015-11-27修回日期:2016-03-11)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
