基于HPLC-MS檢測食品中新霉素和潮霉素B的殘留量
2016-08-02 05:02:29祖立青樂麗歡
山西農(nóng)經(jīng) 2016年18期
關(guān)鍵詞:檢測
□戴 輝 孟 晶 祖立青 樂麗歡
(上海科茂糧油食品質(zhì)量檢測有限公司 上海 普陀區(qū) 200333)
基于HPLC-MS檢測食品中新霉素和潮霉素B的殘留量
□戴 輝 孟 晶 祖立青 樂麗歡
(上海科茂糧油食品質(zhì)量檢測有限公司 上海 普陀區(qū) 200333)
目的建立測定食品中新霉素和潮霉素B氨基糖苷類藥物殘留量的液相色譜-串聯(lián)質(zhì)譜法(HPLC-MS/MS)。方法樣品中的氨基糖苷類藥物經(jīng)磷酸鹽緩沖液提取后,經(jīng)過弱陽離子固相萃取柱富集凈化后,采用液相色譜-三重四極桿串聯(lián)質(zhì)譜檢測,使用電噴霧離子化源(ESI),在正離子條件下以多反應(yīng)監(jiān)測(MRM)方式進(jìn)行掃描。結(jié)果線性范圍為10~1 000μg·kg-1,新霉素和潮霉素B的最低定量濃度(LOQ)為100μg/kg,最低檢測濃度(LOD)為30μg/kg。;以加標(biāo)樣品計(jì)算,在各濃度水平下,各被測物的方法回收率為60%~120%。結(jié)論本方法準(zhǔn)確、高效,適用于食品中氨基糖苷類藥物殘留量的檢測。
氨基糖苷;液相色譜-串聯(lián)質(zhì)譜;殘留檢測
氨基糖苷類化合物(aminoglycosides)作為廣譜抗生素的一種,其抗菌活性是針對諸多革蘭氏陰性菌和革蘭氏陽性菌。氨基糖苷酸抗生素在獸醫(yī)界和畜牧業(yè)中,被廣泛用于治療細(xì)菌性感染。但任何事物都具有兩面性,氨基糖苷類抗生素具有不可逆的耳毒性和腎毒性等不良反應(yīng),為了保證廣大消費(fèi)者安全健康的飲食,美國食品及藥物管理局 (Food and Drug Administration)和歐盟委員會(huì)針對肉類,奶類及奶制品、蛋類等動(dòng)物源性食品進(jìn)行了幾種氨基糖類化合物最大殘留限量(Maximum residuelimit)的設(shè)定。
氨基糖苷類是由兩個(gè)或多個(gè)氨基糖通過苷鍵與氨基環(huán)醇鍵合而成的一類化合物,其結(jié)構(gòu)由于缺少發(fā)色團(tuán)和熒光團(tuán),需要在柱前或柱后衍生化反應(yīng)結(jié)束后,采用液相色譜-熒光或紫外檢測。因?yàn)榘被擒疹惥哂休^強(qiáng)的極性,不易揮發(fā),在進(jìn)行衍生化反應(yīng)后,也會(huì)需要繼續(xù)使用氣相色譜法測定。這些衍生化的方法不僅操作比較繁瑣,而且穩(wěn)定性較差。本次試驗(yàn)采用內(nèi)標(biāo)法,對動(dòng)物源性食品中的新霉素和潮霉素B,這2種氨基糖苷類藥物殘留量進(jìn)行檢測,經(jīng)過較為嚴(yán)格的方法學(xué)驗(yàn)證,對于測定動(dòng)物源食品中氨基糖苷類藥物的殘留量內(nèi)定法具有準(zhǔn)確且高效的特點(diǎn)。
1 材料與方法
1.1 儀器與藥品
LC-MS 8050型三重四極桿串聯(lián)質(zhì)譜儀 (日本島津公司),配有電噴霧離子化源(ESI源);LC-30A超高效液相色譜儀(日本島津公司);MS 3 basic旋渦混合器 (德國 IKA);D-37520臺(tái)式冷凍離心機(jī) (美國Thermo公司)。Oasis混合型弱陽離子(WCX)固相萃取柱(3cm3,60mg,30μm,美國 Waters公司)。
新霉素和潮霉素B對照品購買于美國 Sigma公司;乙腈、甲醇、醋酸、醋酸銨、甲酸均為色譜純;磷酸氫二鈉為分析純;試驗(yàn)用水為超純水。
1.2 方法
1.2.1 試樣制備。肌肉組織及器臟組織、水產(chǎn)品類取適量新鮮或冷藏的樣品,絞碎,并均質(zhì)。置-18℃以下冰箱中冷凍保存。另準(zhǔn)備空白樣品,絞碎后置于-18℃以下冰箱中冷凍保存,作為空白樣品備用。
1.2.2 提取和凈化。前處理1:取5.00g樣品,加入7.5%EDTA緩沖鹽溶液0.5ml(視情況而定,可不加),加入5%三氯乙酸溶液10ml,渦旋1min,超聲20min,4000r/min離心5min,取上清液于一離心管,殘?jiān)屑尤?0ml5%三氯乙酸溶液重復(fù)提取一次,合并上清液。取10ml上清液,用氨水準(zhǔn)確調(diào)ph=7,(用高速離心機(jī)離心后)準(zhǔn)備上WCX小柱。WCX小柱依次用2ml甲醇、2ml水活化。上樣,待樣液自然流完,依次用2ml水,2ml甲醇淋洗,棄去淋洗液,用2ml5%甲酸甲醇洗脫。洗脫液過有機(jī)濾膜后直接上儀器待測。
前處理2:取5.00g樣品,加入7.5%EDTA緩沖鹽溶液0.5ml(視情況而定,可不加),加入5%三氯乙酸溶液10ml,渦旋1min,超聲20min,4000r/min離心5min,取上清液于一離心管,殘?jiān)屑尤?0ml5%三氯乙酸溶液重復(fù)提取一次,合并上清液,取10ml提取液(PH≤1),準(zhǔn)備上HLB小柱。HLB小柱依次用2ml甲醇、2ml水活化。上樣,待樣液自然流完,用5ml水淋洗,2ml甲醇洗脫。洗脫液過濾膜上儀器待測。
前處理3:取5.00g樣品,加入7.5%EDTA緩沖鹽溶液0.5ml(視情況而定,可不加),加入5%三氯乙酸溶液10ml,渦旋1min,超聲20min,4000r/min離心5min,取上清液于一離心管,殘?jiān)屑尤?0ml5%三氯乙酸溶液重復(fù)提取一次,合并上清液,取10ml提取液,準(zhǔn)備上MCX小柱。MCX小柱依次用5ml甲醇、5ml水活化。上樣,待樣液自然流完,依次用5ml水,5ml乙腈,5ml乙腈甲酸銨水溶液(乙腈:2M甲酸銨水溶液=3:7)淋洗,再用5ml乙腈甲酸銨水溶液(乙腈:2M甲酸銨水溶液=3:7)(PH=8.0)洗脫,洗脫液過濾膜上儀器待測。
前處理1可測:壯觀霉素、二氫鏈霉素、潮霉素B、鏈霉素、丁胺卡那霉素、卡那霉素
前處理2可測:安普霉素、妥布霉素、慶大霉素
前處理3可測:新霉素
1.2.3 標(biāo)準(zhǔn)曲線的制備。精密量取10μg/mL10種氨基糖苷類藥物混合標(biāo)準(zhǔn)工作液適量,用樣品空白基質(zhì)配制成氨基糖苷類藥物 濃度為 20、50、100、200、500ng/mL系列標(biāo)準(zhǔn)溶液,供液相色譜-串聯(lián)質(zhì)譜測定。以特征離子質(zhì)量色譜峰面積為縱坐標(biāo),標(biāo)準(zhǔn)溶液濃度為橫坐標(biāo),繪制標(biāo)準(zhǔn)曲線。求回歸方程和相關(guān)系數(shù)。
1.2.4 測定。
(1)色譜條件
a.除新霉素外九種氨基糖苷類
色譜柱:CORTECS Hilic 2.1*100mm 1.6μm;
流動(dòng)相:A:250mM甲酸銨水(200mL中加800uL甲酸)B:200mM甲酸銨有機(jī)相(乙腈+甲醇+水=6+1+3)溶液,(200ml中加甲酸 400uL);
流速:0.25mL/min;
柱溫:30℃;
進(jìn)樣量:5μl;
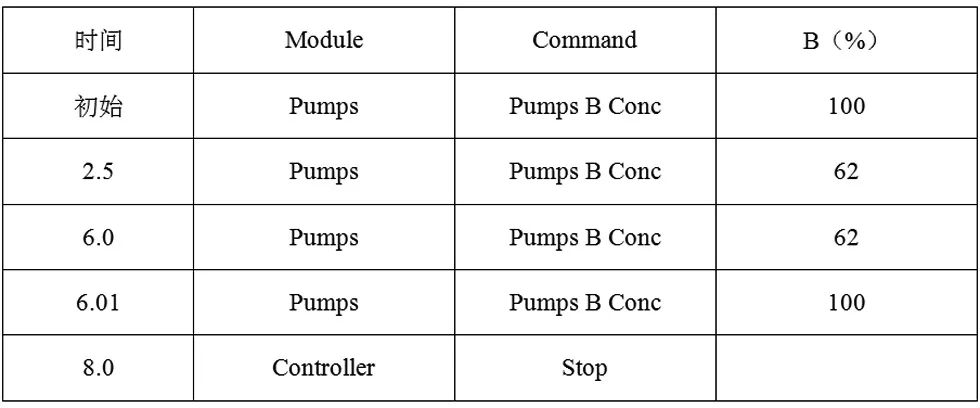
表1 9種氨基糖苷類流動(dòng)相梯度
b.新霉素的色譜條件
色譜柱:ZIC HILLIC 2.1*75mm 3μm;
流動(dòng)相:A:250mM甲酸銨水(200mL中加800uL甲酸)B:200mM甲酸銨有機(jī)相(乙腈+甲醇+水=6+1+3)溶液,(200ml中加甲酸 400uL);
流速:0.25mL/min;
柱溫:30℃;
進(jìn)樣量:5μl;
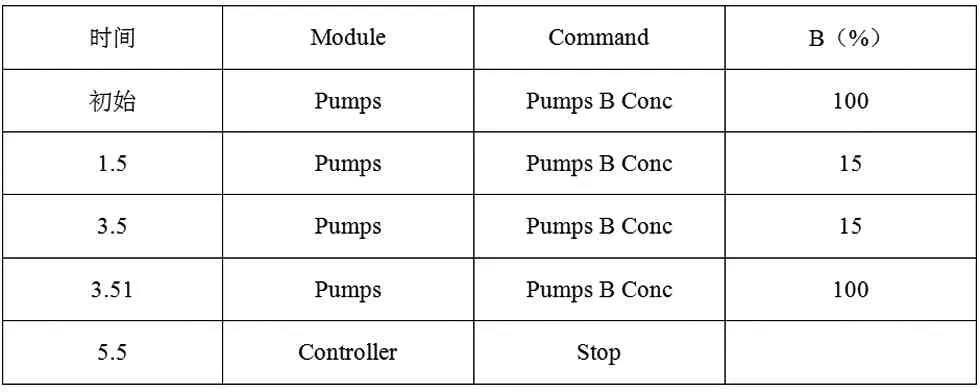
表2 新霉素流動(dòng)相梯度
(2)質(zhì)譜條件
a)離子源:電噴霧離子源;
b)掃描方式:正離子掃描;
c)檢測方式:多反應(yīng)監(jiān)測;
d)毛細(xì)管電壓:Auto Tune(4.0KV)
e)Nebulizing Gas Flow:3L/min;
f)Heating Gas Flow:10L/min;
g)Interface Temperature:300℃;
h)DL Temperature:240℃;
i)Heat Block Temperature:400℃;
j)Drying Gas Flow:10L/min;
k)定量,定性離子對及碰撞能量見表2
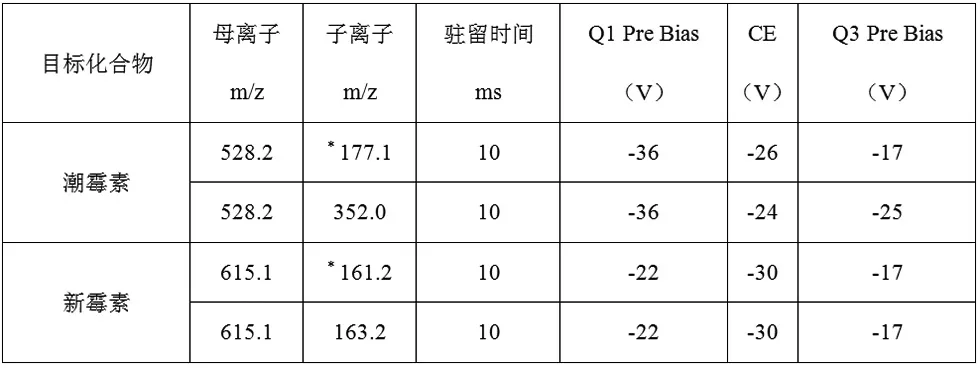
表3 2種氨基糖苷類定量,定性離子對及碰撞能量
1.2.5 空白試驗(yàn)。除不加試料外,采用完全相同的步驟進(jìn)行平行操作。
1.3 計(jì)算方法
1.3.1 定性判斷。在相同實(shí)驗(yàn)條件下,樣品中目標(biāo)化合物與氨基糖苷類標(biāo)準(zhǔn)物質(zhì)的保留時(shí)間一致,且監(jiān)測離子對的相對豐度滿足表3要求。可認(rèn)為目標(biāo)峰是氨基糖苷類物質(zhì)。

表4 目標(biāo)物離子對相對豐度比允許偏差范圍
1.3.2 定量方法。將樣品制備液與標(biāo)準(zhǔn)溶液等體積進(jìn)樣,建立標(biāo)準(zhǔn)工作曲線進(jìn)行定量校準(zhǔn),標(biāo)準(zhǔn)溶液和樣液中待分析物的響應(yīng)均應(yīng)在儀器測定的線性范圍內(nèi)。
計(jì)算公式:
X-樣品中藥物含量(μg/kg);
C-儀器測定樣液中藥物含量(ng/mL);
V-定容體積(mL);
m-稱取樣品質(zhì)量(g);
計(jì)算結(jié)果需扣除空白值,測定結(jié)果用平行測定的算術(shù)平均值表示。
3 結(jié)果
3.1 檢出限
最低定量濃度 (LOQ)與最低檢測濃度(LOD)以信噪比 10∶1和 3∶1分別計(jì)算各被測物的LOQ和 LOD。新霉素和潮霉素B的最低定量濃度(LOQ)為 100μg/kg,最低檢測濃度(LOD)為30μg/kg。
3.2 準(zhǔn)確度
本方法在添加濃度水平為30~200μg/kg時(shí)新霉素和潮霉素B的回收率范圍為60%~120%。
3.3 精密度
本方法的批內(nèi)變異系數(shù)≤20%。
[1]何江,羅先錕,張建宇,李斌.QuEChERS-HPLC-MS/MS在動(dòng)物源食品獸藥多殘留檢測中的研究進(jìn)展[J].廣州化工,2015,(23):56-58.
[2]張林田,黃少玉,陸奕娜,張冬輝,楊晞,孫鋒偉. 固相萃取/HPLC-MS/MS檢測食品中嗎啡等五種罌粟殼生物堿殘留[J].分析試驗(yàn)室,2014,(06):722-725.
[3]楊濤.GPC-HPLC-MS/MS法同時(shí)檢測動(dòng)物源食品中3種獸藥殘留[J].廣州化工,2013,(14):142-144.
[4]劉曉霞.動(dòng)物源性食品中氨基糖苷類抗生素殘留分析方法研究[D].湖南師范大學(xué),2011.
[5]李立,付建,初玉圣,婁喜山,王杰.食品中呋蟲胺殘留量的HPLC-MS/MS檢測方法研究 [J].食品科學(xué),2008,(11):538-540.
1004-7026(2016)18-0097-03
S859.84
A
DOI:10.16675/j.cnki.cn14-1065/f.2016.18.075
戴輝(1985-),男,湖北黃岡人,本科,主要研究方向?yàn)槭称窓z測分析。
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
