代謝工程改造谷氨酸棒桿菌生成丙酮酸
2016-09-14 09:29:23于瑩許湄雪劉金雷范榮馮惠勇李天明
生物技術通報 2016年8期
于瑩 許湄雪 劉金雷 范榮 馮惠勇 李天明
(河北科技大學,石家莊 050018)
代謝工程改造谷氨酸棒桿菌生成丙酮酸
于瑩 許湄雪 劉金雷 范榮 馮惠勇 李天明
(河北科技大學,石家莊 050018)
旨在增加谷氨酸棒桿菌代謝過程中丙酮酸的積累、減少副產物的生成,利用同源重組的方法,敲除了丙酮酸代謝過程中支流代謝途徑的關鍵基因:丙酮酸醌氧化還原酶pqo基因、丙酮酸脫氫酶pdh基因和乳酸脫氫酶lldh基因,獲得了基因缺失工程菌株。利用4.5%的葡萄糖復合培養基,工程菌經搖瓶發酵48 h,丙酮酸的濃度達到14.6 g/L,而野生菌株僅為0.45 g/L;工程菌的糖酸轉化率為33.18%,比野生菌提高了32.5倍。
丙酮酸;谷氨酸棒狀桿菌;丙酮酸醌氧化還原酶;丙酮酸脫氫酶;乳酸脫氫酶
丙酮酸不僅是生物體內糖代謝途徑中重要的有機酸之一,而且也是合成多種化合物的前體,廣泛應用于食品、農藥、生化等工業中[1]。其中,丙酮酸作為酸味添加劑在食品工業中具有很大的發展潛力。丙酮酸是合成丙酮酸鈣的主要原料,丙酮酸鈣有減肥的特效也容易被人體吸收,因此,在食品中添加丙酮酸鈣受到廣大消費者的青睞。隨著對丙酮酸的需求量不斷擴大,對丙酮酸的研究具有十分重要的意義。
生產丙酮酸的方法主要有化學合成法,酶轉化法和微生物發酵法[2]。由于發酵法具有產品純度高、成本低、轉化率高、對環境污染小等優點,已成為國內外最受青睞的生產方法。自然界中有很多的微生物可以合成丙酮酸,如:球擬酵母、假絲酵母、釀酒酵母、腸桿菌屬等。代謝工程可以有效地改造微生物的代謝途徑,從而獲得優良高產菌株。目前,已有文獻[3]報導利用代謝工程技術對大腸桿菌和酵母進行定向改造獲得丙酮酸高產菌株。
谷氨酸棒桿菌(Corynebacterium glutamicum)作為一種重要的工業生產菌,被廣泛應用于氨基酸、有機酸、糖醇的發酵生產,是典型的安全模式菌[4-13]。在谷氨酸棒桿菌代謝過程中,葡萄糖經糖酵解途徑生成磷酸烯醇式丙酮酸,磷酸烯醇式丙酮酸在丙酮酸激酶的作用下生成丙酮酸,丙酮酸在碳源和能量代謝途徑中具有關鍵節點的作用,是合成多種氨基酸和有機酸的前體(圖1)。Blombach等[14]發現缺失丙酮酸脫氫酶pdh基因的谷氨酸棒桿菌相比于野生型,其胞內的丙酮酸含量具有明顯升高;在進一步的研究中,Blombach等[15]發現通過抑制丙酮酸醌氧化還原酶pqo基因能夠影響L-纈氨酸的產量,進而提高了丙酮酸的積累,這些研究為實現細胞內大量積累丙酮酸提供了方向。
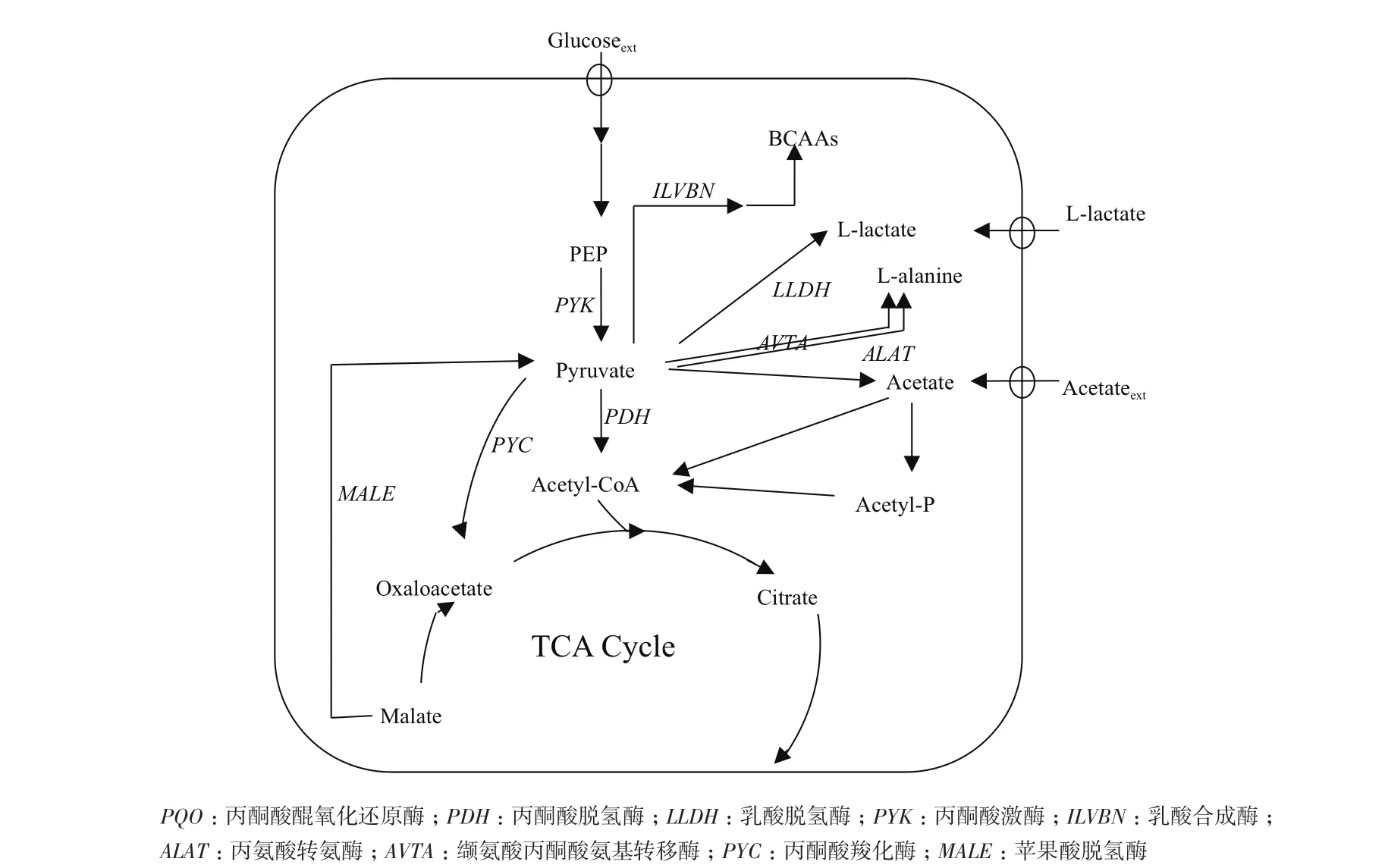
圖1 Corynebacterium glutamicum ATCC 13032代謝途徑
本研究利用兩次同源重組方法和蔗糖致死基因sacB反向篩選技術,敲除控制丙酮酸代謝過程中支流代謝途徑的關鍵基因:丙酮酸醌氧化還原酶pqo基因、丙酮酸脫氫酶pdh基因和乳酸脫氫酶lldh基因[16-19],以期遏制由丙酮酸生成乙酸、乙酰輔酶A 和L-乳酸的支路代謝途徑,使丙酮酸產量得到積累,減少副產物的生成,從而為谷氨酸棒桿菌工業化生產丙酮酸奠定基礎。
1 材料與方法
1.1材料
1.1.1菌株與質粒 本實驗所用的菌株、質粒見表1。
1.1.2主要試劑 High-Fidelity DNA 聚合酶購自北京全式金生物技術有限公司;限制性內切酶、T4連接酶購自NEB公司;質粒提取試劑盒、DNA回收試劑盒購自天根生化科技(北京)有限公司;PCR引物由上海英濰捷基生物技術有限公司合成;其他試劑均為國產分析純,購自生工生物(上海)股份有限公司。
1.1.3儀器與設備 PCR 擴增儀,Eppendorf Mastercycler gradient;全自動凝膠成像系統,Bio-Rad Molecular Imager Gel DOC XR;臺式高速離心機,Eppe-ndorf Mini Spin;高速冷凍離心機,Thermo Sorvall Evolution RC;酶標儀,Bioteke Powerwave;高效液相色譜儀,Agilent 1260 Infinity。

表1 本實驗所用的菌株和質粒及寡核苷酸
1.1.4培養基 LB培養基:1%胰蛋白胨,0.5%酵母提取物,1%氯化鈉,調pH至7.0,固體培養基需加2.2%的瓊脂。
發酵培養基:4.5%葡萄糖,0.5%乙酸鈉,0.1%NHNO3,0.3%(NH4)2SO4,0.1% KH2PO4,0.05% MgSO4,0.02% CaCl2,0.001% FeSO4,0.001% MnSO4,0.0002% CuSO4,0.0001% ZnSO4,0.0002%生物素。
1.2方法
1.2.1基因敲除打靶質粒的構建 pqo基因敲除自殺質粒構建方法為:以谷氨酸棒桿菌基因組為模板,利用PCR擴增出含有pqo基因的上、下同源臂片段,通過Overlapping PCR使 pqo基因的上、下同源臂基因融合,然后將所獲得的基因片段純化后與pK18mobsacB空質粒經HindⅢ和NdeⅠ雙酶切后,將同源臂基因片段連接到質粒上,獲得自殺質粒pK18mobsacB△pqo。本研究用于pdh基因敲除的自殺質粒pK18mobsacB△pdh和用于lldh基因敲除的自殺質粒pK18mobsacB△lldh利用類似方法獲得。
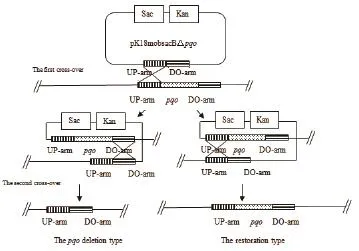
圖2 同源交換原理示意圖
1.2.2敲除系統用于谷氨酸棒桿菌基因的敲除 谷氨酸棒桿菌的基因敲除是利用負篩選標記通過兩次同源重組的方法來實現的,原理示意圖如圖2所示。以獲得突變菌株C. glutamicum△pqo為例,首先將pK18mobsacB△pqo經電轉化轉入野生型谷氨酸棒桿菌感受態細胞中,用含有卡那霉素的LB固體培養基30℃恒溫培養48-60 h,經菌落PCR驗證篩選出第一次同源重組的工程菌,即pK18mobsacB△pqo經同源重組整合到染色體上。再將其接到LB液體培養基中,搖床振蕩自由培養12 h后,涂布于含有10%蔗糖的LB固體培養基上,利用蔗糖致死基因sacB負篩選出第二次同源重組的轉化子,將其一一對應分別擴培到含10%蔗糖和30.0 μg/mL Kan抗性的LB固體培養基中,培養1 d左右。最后挑選在蔗糖板上生長而Kan板上不長的菌落,利用菌落PCR驗證篩選出陽性工程菌C. glutamicum△pqo。利用此方法將pK18mobsacB△pdh轉入C. glutamicum△pqo中,獲得C. glutamicum△pqo△pdh。將pK18mobsacB△lldh轉入C. glutamicum△pqo△pdh中,獲得C. glutamicum△pqo△pdh△lldh。
1.2.3基因缺失突變菌株生長情況驗證 將驗證正確的工程菌C. glutamicum△pqo、C. glutamicum△pqo △pdh、C. glutamicum△pqo△pdh△lldh和野生型菌株接種到搖瓶中作種子過夜培養,后收集菌體,各株菌分別接種到含1.5%葡萄糖的LB培養基、含1.5%葡萄糖和0.5%乙酸鈉的LB培養基中,同時搖瓶振蕩培養,而后通過測定OD值,定期記錄菌株生長情況。
1.2.4丙酮酸的發酵方法 從LB固體培養基上挑取工程菌C. glutamicum△pqo、C. glutamicum△pqo △pdh、C. glutamicum△pqo△pdh△lldh與野生型菌株,分別接種到LB培養基中作種子,30℃、180 r/min培養過夜,后將培養物離心收集菌體,轉移到含有發酵培養基的三角瓶中,以4.5%的葡萄糖作為碳源,相同條件繼續搖床振蕩培養,發酵48 h,整個發酵過程中定時調節pH,使之維持在7.0。
1.2.5高效液相色譜檢測丙酮酸的含量 將待測發酵液經轉速8 000 r/min離心10 min,取上清液經0.22 μm醋酸纖維素濾膜過濾后,利用高效液相色譜法分析丙酮酸的含量。檢測條件為:美國伯樂Aminex HPX-87H Column,流動相0.005 mol/L H2SO4,檢測波長210 nm,流速0.6 mL/min,柱溫55℃,進樣量20 μL。
2 結果
2.1基因敲除打靶質粒的構建與驗證
利用Overlapping PCR將目的基因的上、下同源臂基因片段融合搭接,后與pK18mobsacB空質粒同時雙酶切,獲得用于基因打靶的重組質粒,而后通過雙酶切鑒定上下同源臂是否連接到載體上。重組質粒pK18mobsacB△pqo的雙酶切驗證結果(圖3-A)顯示,經限制性內切酶酶切后,分別得到大小為5 400 bp的質粒和大小為2 000 bp的同源臂片段,與理論值一致,表明pqo基因的上、下同源臂基因連接至pK18mobsacB空質粒。重組質粒pK18mobsacB△pdh的雙酶切驗證結果(圖3-B)顯示,經限制性內切酶酶切后,分別得到大小為5 400 bp的質粒和大小為2 200 bp的同源臂片段,與理論值一致,表明pdh基因的上、下同源臂基因連接至pK18mobsacB空質粒。重組質粒pK18mobsacB△lldh的雙酶切驗證結果(圖3-C)顯示,經限制性內切酶酶切后,分別得到大小為5 400 bp的質粒和大小為1 800 bp的同源臂片段,與理論值一致,表明lldh基因的上、下同源臂基因連接至pK18mobsacB空質粒。
2.2谷氨酸棒桿菌基因敲除菌株的驗證
由圖2同源重組示意圖可知,谷氨酸棒桿菌經過兩次同源重組,能夠出現一種情況是基因缺失突變菌株,另一種情況是基因恢復菌株,因而需要通過PCR對發生第2次同源重組的突變菌株進行驗證。將打靶質粒pK18mobsacB△pqo經電轉化轉入野生型谷氨酸棒桿菌感受態細胞中,用含有卡那霉素的固體LB培養基篩選得到第一次同源重組的工程菌,再利用蔗糖致死基因sacB基因負篩選出第二次同源重組的轉化子,最后得到C. glutamicum△pqo。通過NCBI查找谷氨酸棒桿菌ATCC13032基因組序列中pqo基因序列,設計敲除驗證引物,分別以基因敲除菌株C. glutamicum△pqo與野生菌株的基因組DNA為模板,進行PCR驗證,結果(圖4-A)顯示,野生菌擴增出大小為850 bp的pqo基因片段,而C. glutamicum△pqo菌株沒有擴增片段,說明C. glutamicum△pqo工程菌株基因組中的pqo基因已被敲除。同樣,將pK18mobsacB△pdh轉入C. glutamicum△pqo中,獲得C. glutamicum△pqo△pdh,分別以C. glutamicum△pqo和C. glutamicum△pqo△pdh為模板,PCR驗證pdh基因片段,結果如圖4-B所示。將pK18mobsacB△lldh轉入C. glutamicum△pqo△pdh中,獲得C. glutamicum△pqo△pdh△lldh,并分別以C. glutamicum△pqo△pdh和C. glutamicum△pqo△pdh △lldh為模板,PCR驗證lldh基因片段,結果如圖4-C所示。
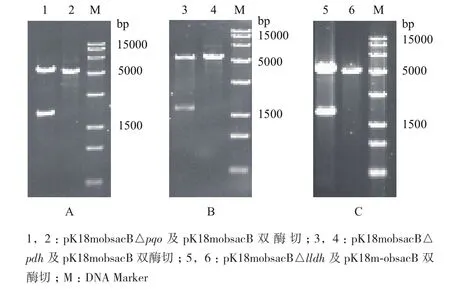
圖3 重組質粒pK18mobsacB△pqo(A)、pK18mobsacB △pdh(B)、pK18mobsacB△lldh(C)的酶切驗證
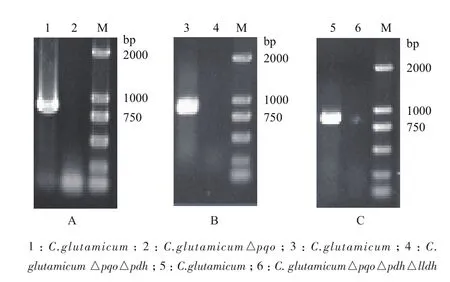
圖4 工程菌株C. glutamicum△pqo(A)、C. glutamicum △pqo△pdh(B)、C. glutamicum△pqo△pdh△lldh (C)PCR驗證
2.3工程菌生長特性研究
C. glutamicum△pqo、C. glutamicum△pqo△pdh、C. glutamicum△pqo△pdh△lldh與野生菌以等OD值分別接種到含1.5%葡萄糖的LB培養基、含1.5%葡萄糖和0.5%乙酸鈉的LB培養基中。生長狀況如圖5所示,生長曲線(圖5-A)顯示在不含乙酸的培養基中,野生型C. glutamicum和C. glutamicum △pqo生長,而敲除了pdh基因的C. glutamicum△pqo△pdh和C. glutamicum△pqo△pdh△lldh無法正常生長;生長曲線(圖5-B)顯示在含乙酸的培養基中,由于乙酸的補給,使pdh基因缺失菌株C. glutamicum△pqo △pdh和C. glutamicum△pqo△pdh△lldh與野生型C. glutamicum和C. glutamicum △pqo菌株生長的趨勢一致。這就說明由于敲除了pdh基因,使丙酮酸脫氫酶失活,遏制了丙酮酸生成乙酰輔酶A,影響了代謝途徑的TCA循環,只有在培養基中添加0.5%乙酸鈉才能正常生長,進而驗證了乙酸鈉是工程菌能夠正常生長所必需的碳源。
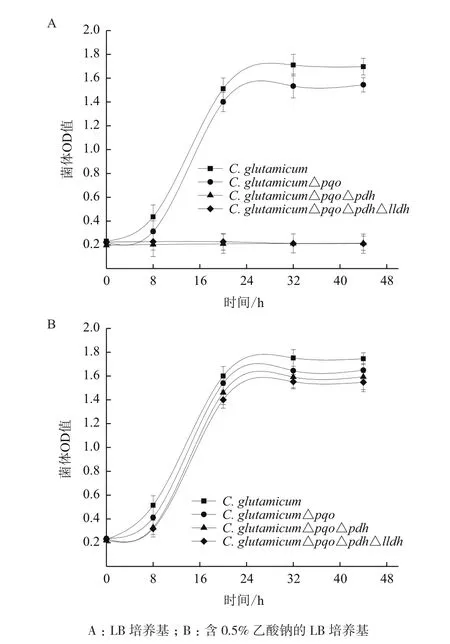
圖5 野生菌與工程菌的生長情況
2.4工程菌丙酮酸的發酵
為了進一步驗證重組菌種中丙酮酸產量,分別將C. glutamicum△pqo、C. glutamicum△pqo△pdh、C. glutamicum△pqo△pdh△lldh與野生菌C. glutamicum同時以4.5%的葡萄糖作為碳源進行搖瓶發酵,48 h后收集發酵液,利用高效液相色譜法測定發酵液中丙酮酸的含量。結果(圖6)顯示,由于工程菌C. glutamicum△pqo△pdh△lldh敲除了丙酮酸代謝過程中的3個關鍵基因,遏制了副產物的形成,因而積累的丙酮酸含量要明顯高于野生型菌株、僅敲除一個基因的C. glutamicum△pqo和敲除兩個基因的C. glutamicum△pqo△pdh。經計算可知野生型菌株發酵后丙酮酸含量僅為0.45 g/L,而工程菌C. glutamicum△pqo△pdh△lldh發酵后丙酮酸含量為14.6 g/L,工程菌比野生菌提高了31.4倍;工程菌株的糖酸轉化率為33.18%,比野生菌提高了32.5倍。
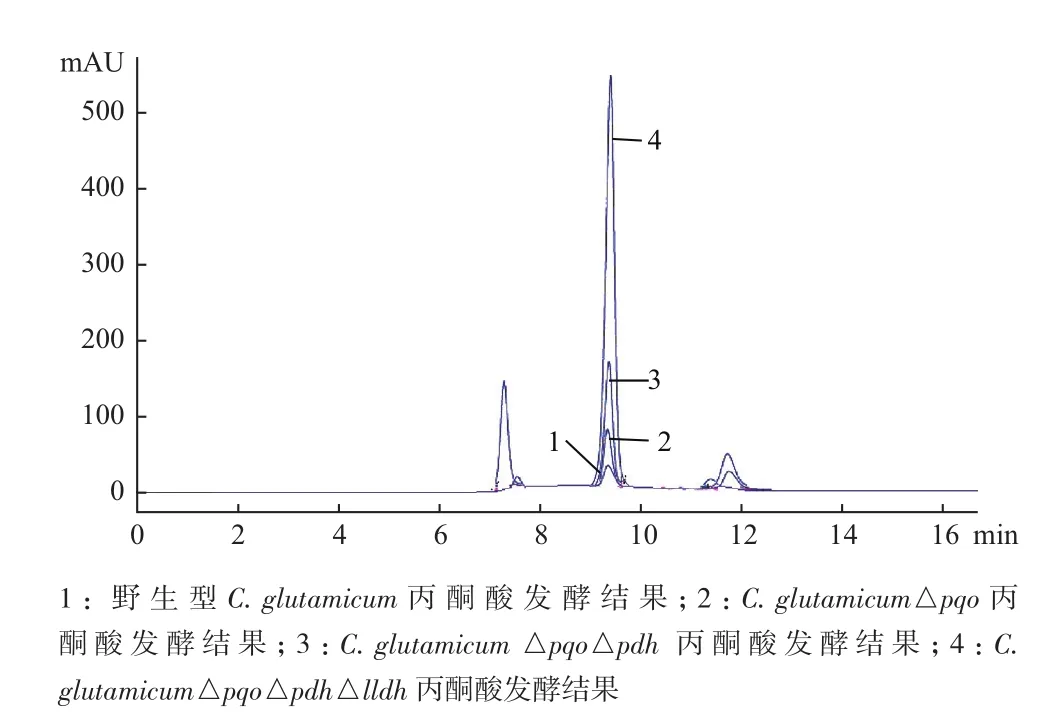
圖6 野生菌與工程菌丙酮酸發酵液相對比圖
3 討論
傳統的發酵法生產丙酮酸,產量和糖酸轉化率較低,遠遠不能滿足日益增加的市場需求。近年來應用缺陷性酵母菌和大腸桿菌來生產丙酮酸成為人們研究的熱點。Li等[20]利用酵母發酵生產丙酮酸,通過復雜的發酵設置維持培養基中維生素的濃度來提高丙酮酸的產量;Zelic等[21]利用大腸桿菌工程菌進行搖瓶發酵,能夠達到每摩爾葡萄糖生產1.9 mol的丙酮酸;Zhu等[22]改造的用于丙酮酸生產的大腸桿菌,在最優發酵條件下丙酮酸產量能達到90 g/L。而谷氨酸棒桿菌是食品級微生物、魯棒性好,更適合工業生產條件,利用的代謝工程改造谷氨酸棒桿菌生產丙酮酸,目前還未見報道。本研究根據谷氨酸棒桿菌的代謝途徑設計策略,敲除pqo基因使其以葡萄糖為碳源不能生成乙酸;以pqo基因缺失的工程菌C. glutamicum△pqo為基礎菌株敲除編碼丙酮酸脫氫酶pdh基因,有效的阻止了乙酰輔酶A的生成,影響了TCA循環,只有在含乙酸鈉的培養基中才能夠穩定生長;敲除編碼乳酸脫氫酶的lldh基因,減少了副產物乳酸的生成,從而提高了丙酮酸的積累量。獲得的基因缺失工程菌C. glutamicum △pqo△pdh△lldh,以4.5%的葡萄糖為碳源,進行搖瓶發酵,48 h后,工程菌中丙酮酸的濃度為14.6 g/L,比野生菌株提高了14.15 g/L;工程菌的糖酸轉化率為33.18%,比野生菌提高了32.5倍,生產效率為0.3 g/(L·h)。具Zhu[22]和Li[20]的報道,大腸桿菌工程菌發酵罐發酵的生產率為1.23 g/(L·h),酵母菌發酵罐發酵的生產率為1.875 g/(L·h)。與大腸桿菌工程菌和酵母菌工程菌相比,本研究還有一定差距,一是因為搖瓶發酵條件沒有發酵罐的優越,二是因為丙酮酸形成后通過合成纈氨酸、丙氨酸等支流代謝丙酮酸分解消耗,如編碼生成乙酰乳酸的乳酸合成酶ilvbn基因、編碼生成L-丙氨酸的丙氨酸轉氨酶alat基因和纈氨酸丙酮酸氨基轉移酶avta基因、編碼生成草酰乙酸的丙酮酸羧化酶pyc基因等,影響了丙酮酸的積累。另外,強化丙酮酸合成代謝的基因,如編碼蘋果酸脫氫酶male基因將蘋果酸代謝生成丙酮酸等,將有利于丙酮酸的積累,提高丙酮酸的產量。
4 結論
本研究利用兩次同源重組方法和蔗糖致死基因sacB反向篩選技術,敲除了控制丙酮酸代謝過程中支流代謝途徑的關鍵基因,獲得基因缺失工程菌C. glutamicum △pqo△pdh△lldh,進而遏制了由丙酮酸生成乙酸、乙酰輔酶A和L-乳酸的支路代謝途徑,使丙酮酸積累量大大增加,以4.5%的葡萄糖為碳源,進行搖瓶發酵,48 h后,工程菌中丙酮酸的濃度為14.6 g/L,比野生菌株提高了14.15 g/L;工程菌的糖酸轉化率為33.18%,比野生菌提高了32.5倍,生產效率為0.3 g/(L·h)。
[1]郭英, 付濤. 發酵法生產丙酮酸的工藝條件優化研究[J]. 石家莊職業技術學院學報, 2010, 22(2):26-28.
[2]劉立明, 李寅, 堵國成, 等. 生物技術法生產丙酮酸的研究進展[J]. 生物工程學報, 2002, 18(6):651-655.
[3]Wieschalka S, Blombach B, EikmannsI BJ. Engineering Corynebacterium glutamicum for the production of pyruvate[J]. Applied Microbiology and Biotechnology, 2012, 94:449-459.
[4]吳新陽, 裴廣勝, 等. 不同抑制劑對谷氨酸棒狀桿菌磷酸烯醇式丙酮酸羧化酶酶活的影響[J]. 生物技術通報, 2013(7):172-178.
[5]張君勝, 王揚, 楊曉志, 張力. 谷氨酸棒桿菌關鍵酶活性與蘇氨酸高產的關系[J]. 湖南農業科學, 2011(9):23-25.
[6]Wieschalka S, Blombach B, Bott M, et al. Bio-based production of organic acids with Corynebacterium glutamicum[J]. Microbial Biotechnology, 2012, 6:87-102.
[7]Lin LH, Hu XQ, Xu DQ, et al. Co-expression of feedback-resistant threonine dehydratase and acetohydroxy acid synthase increase L-isoleucine production in Corynebacterium glutamicum[J]. Metabolic Engineering, 2012, 14:542-550.
[8]Rado? D, Turner DL, Fonseca LL, et al. Carbon flux analysis by13C nuclear magnetic resonance to determine the effect of CO2on anaerobic succinate production by Corynebacterium glutamicum[J]. Applied and Environmental Microbiology, 2014,80(10):3015-3024.
[9]Bommareddy RR, Chen Z, Rappert S, et al. A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase[J]. Metabolic Engineering, 2014, 25:30-37.
[10]Vogt M, Haas S, Klaffl S, et al. Pushing product formation to its limit:metabolic engineering of Corynebacterium glutamicum for L-leucine overproduction[J]. Metabolic Engineering, 2014, 22:40-52.
[11]Baumgart M, Unthan S, Rückert C, et al. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology[J]. Applied and Environmental Microbiology,2013, 79(19):6006-6015.
[12]Zhang Y, Shang XL, Lai SHJ, et al. Development and application of an arabinose-inducible expression system by facilitating inducer uptake in Corynebacterium glutamicum[J]. Applied and Environmental Microbiology, 2012, 78(16):5831-5838.
[13]Yamamoto S, Gunji W, Suzuki H, et al. Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions[J]. Applied and Environmental Microbiology, 2012, 78(12):4447-4457.
[14]Blombach B, Schreiner ME, Holátko J, et al. L-valine production with pyruvate dehyrogenase complex-deficient Corynebacterium glutamicum[J]. Appl Environ Microbiol, 2007, 73(7):2079-2084.
[15]Blombach B, Schreiner ME, Bartek T, et al. Corynebacterium glutamicum tailored for high-yield L-valine production[J]. Appl Microbiol Biotechnol, 2008, 79:471-479.
[16] Litsanov B, Brocker M, Bott M. Toward homosuccinate fermentation:metabolic engineering of Corynebacterium glutamicum for anaerobic production of succinate from glucose and formate[J]. Applied and Environmental Microbiology, 2012, 78(9):3325-3337.
[17] Yamamoto S, Gunji W, Suzuki H, et al. Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions[J]. Applied and Environmental Microbiology, 2012, 78(12):4447-4457.
[18]Zahoor A, Lindner SN, Wendisch VF. Metabolic engineering of Corynebacterium glutamicum aimed at alternative carbon sources and new products[J]. Computational and Structural Biotechnology Journal, 2012, 3:1-11.
[19]Litsanov B, Kabus A, Brocker M, et al. Efficient aerobic succinate production from glucose in minimal medium with Corynebacterium glutamicum[J]. Microbial Biotechnology, 2012, 5(1):116-128.
[20]Li Y, Chen J, Lun SY. Biotechnological production of pyruvic acid[J]. Applied Microbiology and Biotechnology, 2001, 57:451-459.
[21]Zeli? B, Gerharz T, Bott M, et al. Fed-batch process for pyruvate production by recombinant Escherichia coli YYC202 strain[J]. Eng in life Sci, 2003, 3:299-305.
[22]Zhu Y, Eiteman MA, Altman R, Altman E. High glycolytic flux improves pyruvate production by a metabolically engineered Escherichia coli strain[J]. Appl Environ Microbiol, 2008, 74(21):6649-6655.
(責任編輯 馬鑫)
Metabolic Engineering for Modifying Corynebacterium glutamicum to Produce More Pyruvate
YU Ying XU Mei-xue LIU Jin-lei FAN Rong FENG Hui-yong LI Tian-ming
(Hebei University of Science and Technology,Shijiazhuang 050018)
In order to increase the accumulation of pyruvate while decrease the production of by-products during the metabolic process of Corynebacterium glutamicum,the homologous recombination was employed to knock out the key genes of pyruvate:quinone oxidoreductase (pqo),pyruvate dehydrogenase(pdh)and L-lactate dehydrogenase(lldh)in the tributary metabolic pathways,thus the gene deletion mutation strains were constructed. Using compound medium with 4.5% glucose as carbon sources,the concentration of pyruvate with mutation strain at shake flask fermentation for 48 hours reached 14.6 g/L,while that with the wild type only was 0.45 g/L. The conversion rate of sugar to acid with the mutation strain was 33.18% and 32.5 times higher than the wild type.
pyruvate;Corynebacterium glutamicum;pyruvate:quinone oxidoreductase;pyruvate dehydrogenase;L-lactate dehydrogenase
10.13560/j.cnki.biotech.bull.1985.2016.08.033
2015-12-08
國家科技支撐計劃(2015BAD15B05)
于瑩,女,碩士研究生,研究方向:代謝工程與合成生物學;E-mail:503572523@qq.com
李天明,男,助理研究員,研究方向:代謝工程與合成生物學;E-mail:iamltm2000@hotmail.com
