不同產地白術藥材中白術內酯I和II的含量測定
2016-09-20 02:09:55吳梓春何兆錦廣東省佛山市第一人民醫院中山大學附屬佛山醫院藥學部佛山528000
北方藥學 2016年9期
吳梓春 何兆錦(廣東省佛山市第一人民醫院(中山大學附屬佛山醫院)藥學部 佛山 528000)
不同產地白術藥材中白術內酯I和II的含量測定
吳梓春何兆錦(廣東省佛山市第一人民醫院(中山大學附屬佛山醫院)藥學部佛山528000)
目的:探討不同產地白術藥材中白術內酯I和白術內酯II的高效液相測定方法。方法:選擇浙江、安徽、甘肅、江西四個產地的白術藥材,色譜柱采用Hypersil 0DS(4.6mm×250mm,2.5μm),流動相為乙腈∶水=55∶45,流速為1.0ml/min,柱溫為30℃,檢測波長223nm。結果:白術內酯I的線性關系方程為:Y=3.546×104+0.524×104(r=0.9998),在0.5~500μg范圍內線性關系良好;白術內酯II的線性關系方程為:Y=4.746×104+0.949×104(r=0.9997),在0.5~500μg范圍內線性關系良好。白術內酯I的平均加樣回收率為99.74%,RSD1.32%,白術內酯II平均加樣回收率為99.68%,RSD為2.01%。浙江產白術藥材中兩種有效成分含量最高,江西產白術藥材的兩種有效成分含量較低。結論:高效液相色譜法測定不同產地白術中的有效成分,方法簡單易操作,靈敏度高,準確性好,重復性強,可以作為白術質量控制的定量指標。
白術內酯I白術內酯II高效液相色譜 不同產地
中藥白術為菊科(Composetae)蒼術屬(Atractylodes)植物白術(Atractylodes macrocephala Koidz)的干燥根莖[1],多數分布于我國的浙江、安徽、甘肅、湖南以及江西等產地[2]。白術味苦、甘,性溫,主要歸脾胃經,功能健脾益氣,燥濕利水,為補益類的補氣中藥材[3]。由于我國白術的產地較多,藥材的質量也有一定的差異,我國各個地域的生態環境、氣候等不同,導致了不同產地白術藥材有效成分的含量存在差異。為了加強白術藥材的質量控制,本研究以白術中白術內酯I、白術內酯II的含量作為觀察指標,采用高效液相色譜法觀察不同產地白術中上述兩種有效成分的含量。
1 儀器與試藥
1.1實驗儀器:日本島津LC-10AT型單泵高效液相色譜儀,SPD-10AVP型紫外檢測器,島津CBM色譜工作站。手提式高速萬能粉碎機DFD-100、4號篩,電熱恒溫鼓風干燥箱,KQ-500E型超聲波清洗儀,美國梅特勒電子分析天平(精密度為0.0001g),PL-80離心沉淀機。
1.2實驗試藥:白術藥材采自浙江、安徽、甘肅、江西四個產地,藥材均經專家鑒定分別為菊科(Compositae)植物白術(Atractylodes macrocephala koidz)的干燥根莖和菊科植物Atractylades macrocephalacv.Yuzhu的干燥根莖。乙腈為色譜純,水為三蒸水,其他試劑均為分析純。
2 方法與結果
2.1色譜條件的選擇:色譜柱采用Hypersil 0DS(4.6mm×250mm,2.5μm),流動相為乙腈∶水=55∶45,流速為1.0ml/min,柱溫為30℃,檢測波長223nm,進樣量為20μL。以白術內酯I作為參考,色譜柱的理論塔板數>5000。
2.2對照品溶液的制備:精密稱定白術內酯I對照品0.4mg,置于5ml容量瓶中,甲醇溶液定溶至刻度后搖勻,靜置;取白術內酯II對照品0.45mg精密稱定后置于5ml容量瓶中,甲醇溶液定溶至刻度后搖勻,靜置。兩種對照品溶液置于-20℃冰箱中保存,備用。
2.3供試品溶液的制備:取白術藥材2.0g置于粉碎機中粉碎后過4號篩,精密稱定后置于加塞錐形瓶中,加入甲醇溶液50ml后稱定重量,超聲處理40min。放冷后再次稱定重量,甲醇溶液將減少的重量補足后以塞蓋住錐形瓶,搖勻,過0.45μm微孔濾膜后,置于3000轉/min的離心機中離心10min,取上清液即得供試品溶液。
2.4標準曲線的繪制:將分別溶解于甲醇溶液中的白術內酯I、白術內酯II對照品溶液稀釋成0.5、1.0、5.0、10.0、20.0、50.0、80.0、100.0μg/ml的白術內酯I、II標準溶液,按照“2.1”項下的色譜條件,分別進樣20μL,以白術內酯I、II色譜峰面積作為縱坐標,白術內酯I、II對照品質量濃度為橫坐標繪制標準曲線,得到回歸方程以及相關系數。白術內酯I的線性關系方程為:Y= 3.546×104+0.524×104(r=0.9998),在0.5~500μg范圍內線性關系良好;白術內酯II的線性關系方程為:Y=4.746×104+0.949×104(r=0.9997),在0.5~500μg范圍內線性關系良好。
2.5穩定性考察:取同一批次的樣品藥材,按照“2.3”項下供試品溶液的制備方法進行操作,制備供試品溶液后精密吸取同一供試品溶液,按照“2.1”項下的色譜條件進樣,分別在配制溶液后0、1、4、6、8、12、24h吸取20μL進樣分析,按照線性關系方程計算白術內酯I、白術內酯II在供試品溶液中的穩定性,結果表明白術內酯I的RSD值為2.14%,白術內酯II的RSD值為2.01%,表明溶液在24h內較為穩定。
2.6精密度考察:將:2.5:項下配制的同一批次供試品溶液,吸取20μL,按照“2.1”項下的色譜條件重復進樣5次,計算峰面積值,結果表明其RSD值為1.36%,表明該儀器的精密度良好。
2.7重現性考察:取同一白術樣品粉末,精密稱取5份,每份為1.245g,按照“2.3”項下的供試品溶液的制備方式進行配制,按照“2.1”項下的色譜條件進樣后得到白術內酯I、白術內酯II的RSD分別為1.84%、1.76%,表明該色譜條件下測定結果的重現性良好。
2.8加樣回收率試驗:精密稱取已知白術內酯I、白術內酯II含量的白術粉末,精密稱定5份,精密加入白術內酯I、II對照品適量,按照“2.3”項下供試品溶液的制備方式進行供試品溶液的配制,按照“2.1”項下的色譜條件進行色譜圖的分析,計算平均加樣回收率,結果見表1、表2。
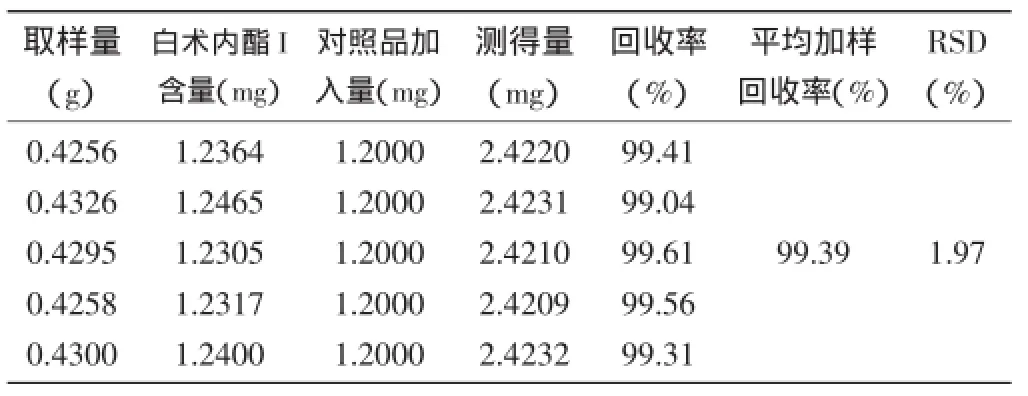
表1 白術內酯I加樣回收率測定結果(n=5)

表2 白術內酯II 加樣回收率測定結果(n=5)
2.9不同產地白術含量測定結果:取浙江、安徽、甘肅、江西白術樣品約1.0g,精密稱定后按照“2.3”項下的供試品溶液制備溶液并進行處理測定,按照“2.1”項下的色譜條件進樣,計算不同產地白術藥材中白術內酯I、II的含量并進行比較,結果見表3。
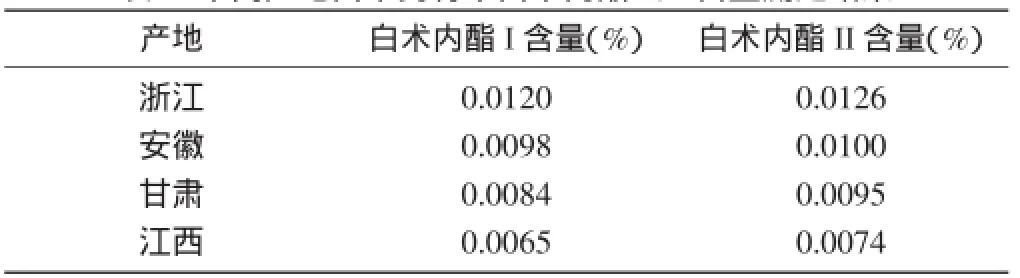
表3 不同產地白術藥材中白術內酯I、II含量測定結果
3 討論
3.1流動相的選擇:本研究中采用了乙腈:水,甲醇:水等按照不同比例流動相系統,結果顯示在乙腈∶水=55∶45時色譜系統可以獲得較好的峰形,同時色譜峰分離較好。因此實驗中選擇乙腈:水作為流動相。
3.2檢測波長的選擇:采用Agilent 1100DAD三維圖譜進行分析得到白術內酯類化合物在223nm處有最大的吸收峰,為了在同一條件下進行檢測,選擇同時檢測兩個波長的信號[4]。
不同產地的白術內酯含量具有一定的差異[5],在相同的測定條件下,分離得到的白術內酯I、II含量,浙江產地的白術藥材中最多,江西產地的藥材則偏少,因此如果要獲得較多含量的白術內酯可以選擇浙江產的白術中藥材。
[1]趙桂芝,洪學智.白術內酯的藥理學研究進展[J].中國藥房,2012,20(3):230-231.
[2]曾志,周育妹.3個不同產地白術的揮發性化學成分比較[J].華南師范大學學報,2012,47(5):78-83.
[3]趙紅紅.三產地白術揮發油氧化分解后化學成分的變化[J].山西大學學報,2015,38(3):516-521.
[4]田穎.不同產地白術藥材中白術內酯II和III的含量測定[J].海峽藥學,2012,24(8):65-67.
[5]姜東京,徐志偉.浙江道地藥材於術與白術的質量對比研究[J].中華中醫藥學刊,2014,32(12):2864-2866.
R284.1
A
1672-8351(2016)09-0010-02
