大孔樹脂分離純化西洋參葉總皂苷的工藝研究△
2016-09-25 08:30:29郭婷婷王兆華張大軍
中國現(xiàn)代中藥 2016年9期
郭婷婷,王兆華,張大軍
(1.長春中醫(yī)藥大學(xué)附屬醫(yī)院,吉林 長春 130021; 2.吉林省中醫(yī)藥科學(xué)院,吉林 長春 130012)
大孔樹脂分離純化西洋參葉總皂苷的工藝研究△
郭婷婷1,王兆華2*,張大軍2
(1.長春中醫(yī)藥大學(xué)附屬醫(yī)院,吉林 長春130021;2.吉林省中醫(yī)藥科學(xué)院,吉林 長春130012)
目的:探索以D101型大孔吸附樹脂分離純化西洋參葉總皂苷的工藝條件及參數(shù)。方法:以總皂苷及人參皂苷Rb3的吸附率和解吸附率為指標(biāo),對大孔樹脂分離純化西洋參葉總皂苷的工藝條件進行考察,并對其吸附動力學(xué)曲線和動態(tài)解析曲線進行探索。結(jié)果:藥材與濕樹脂量比為3∶10,上柱流速為1.5mL·min-1,重復(fù)上樣兩次,分別用3倍體積的水、3倍體積70%乙醇洗脫,收集70%乙醇洗脫液,減壓,濃縮,干燥,即得總皂苷提取物。提取物中總皂苷含量>75%,人參皂苷Rb3含量>10%,總皂苷提取物得率約為14%。結(jié)論:該工藝簡便,重復(fù)性好,可用于西洋參葉中總皂苷的提取純化。
西洋參葉;大孔吸附樹脂;總皂苷;人參皂苷Rb3;工藝參數(shù)
西洋參PanaxquinquefoliusL.為五加科人參屬植物,原產(chǎn)于美國北部到加拿大南部一帶,北京、陜西、吉林等地已引種栽培成功[1]。由于西洋參葉所含的主要活性物質(zhì)—人參皂苷在種類上與主根基本一致,而在含量上明顯高于主根等其他藥用部位[2-3],因而具有較高的開發(fā)利用價值。D101型大孔吸附樹脂分離純化總皂苷是目前國內(nèi)外常用的方法[4],這種方法操作簡單、樹脂可反復(fù)利用、成本低、安全無毒。本實驗系統(tǒng)考察了西洋參葉總皂苷的工藝參數(shù),包括吸附量、吸附流速、脫附性能、藥液濃度等,為西洋參葉總皂苷提取物的制備工藝提供實驗基礎(chǔ)。
1 儀器和試藥
UV-2800紫外-可見分光光度計(上海精密科學(xué)儀器有限公司);日本島津LC-10AT高效液相色譜儀、SPD-10A紫外檢測器(日本島津公司);KQ-250型超聲波處理器(天津奧特賽恩斯儀器公司);AE163電子天平(瑞士梅特勒公司)。
人參皂苷Rb3對照品(中國食品藥品檢定研究院,批號:111686-2010002,供含量測定用);D101大孔吸附樹脂(安徽三星樹脂科技有限公司,藥用級);西洋參葉購自靖宇參園;甲醇為色譜純;乙醇、硫酸、冰醋酸、香草醛等均為分析純。
2 方法與結(jié)果
2.1總皂苷含量測定
2.1.1標(biāo)準(zhǔn)曲線的制備 取人參皂苷Rb3對照品,加甲醇制成質(zhì)量濃度為0.964mg·mL-1的溶液,搖勻。分別精密吸取上述溶液25、50、100、150、200、250μL,分別置于具塞試管中,蒸干,加入1%香草醛高氯酸試液0.5mL,置60℃水浴上加熱15min,立即取出,用冰水冷卻2min,加入77%硫酸溶液5mL,搖勻,以相應(yīng)試劑作為空白。于540nm波長處測定吸收度,以吸光度為縱坐標(biāo),濃度為橫坐標(biāo)繪制標(biāo)準(zhǔn)曲線,線性范圍為24.10~241.0μg。
2.1.2供試品溶液的制備與測定 取西洋參葉總皂苷提取物0.15g,精密稱定,置錐形瓶中,精密加入甲醇50mL,稱定重量,超聲處理30min,放冷,再稱定重量,用甲醇補足減失的重量,濾過,精密量取續(xù)濾液10mL,蒸干,殘渣加水10mL使溶解,以水飽和正丁醇提取3次,每次20mL,合并正丁醇提取液,以正丁醇飽合水溶液洗滌2次,每次15mL,取正丁醇液蒸干,殘渣加甲醇溶解,并轉(zhuǎn)移至10mL量瓶內(nèi),以甲醇稀釋至刻度,搖勻,作為供試品溶液[5]。精密吸取供試品溶液40μL,蒸干,照標(biāo)準(zhǔn)曲線制備項下方法自“加入1%香草醛高氯酸試液0.5mL”起,依法測量吸收度,計算樣品的含量。
2.1.3檢測波長的選擇 取對照品溶液和供試品溶液,顯色后在480~620nm波長范圍內(nèi)掃描,最終確定檢測波長為540nm。光譜掃描曲線見圖1。
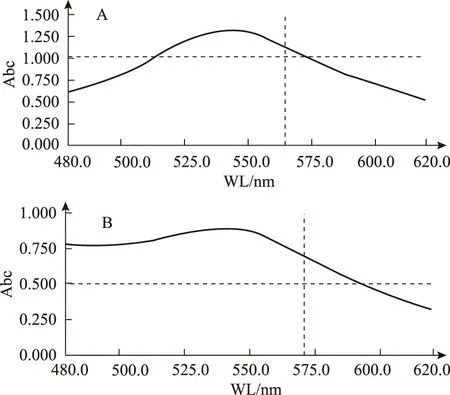
注:A.人參皂苷Rb3對照品;B.西洋參葉供試品。圖1 對照品和供試品光譜掃描曲線
2.2人參皂苷Rb3含量測定
2.2.1 色譜條件 Agilent ZORBAX SB-C18柱(150 mm×4.6 mm,5 μm);以乙腈-水(32︰68)為流動相;檢測波長為203 nm;柱溫為室溫,理論板數(shù)按人參皂苷Rb3計算應(yīng)不低于2500。
2.2.2 對照品溶液的制備 取人參皂苷Rb3對照品適量,精密稱定,加甲醇制成質(zhì)量濃度為 0.5 mg·mL-1的溶液,即得。
2.2.3 供試品溶液的制備與測定 分別精密吸取2.1.2項下供試品溶液及上述對照品溶液各10 μL,按2.2.1項下的色譜條件進行測定,計算含量,液相色譜圖見圖2。
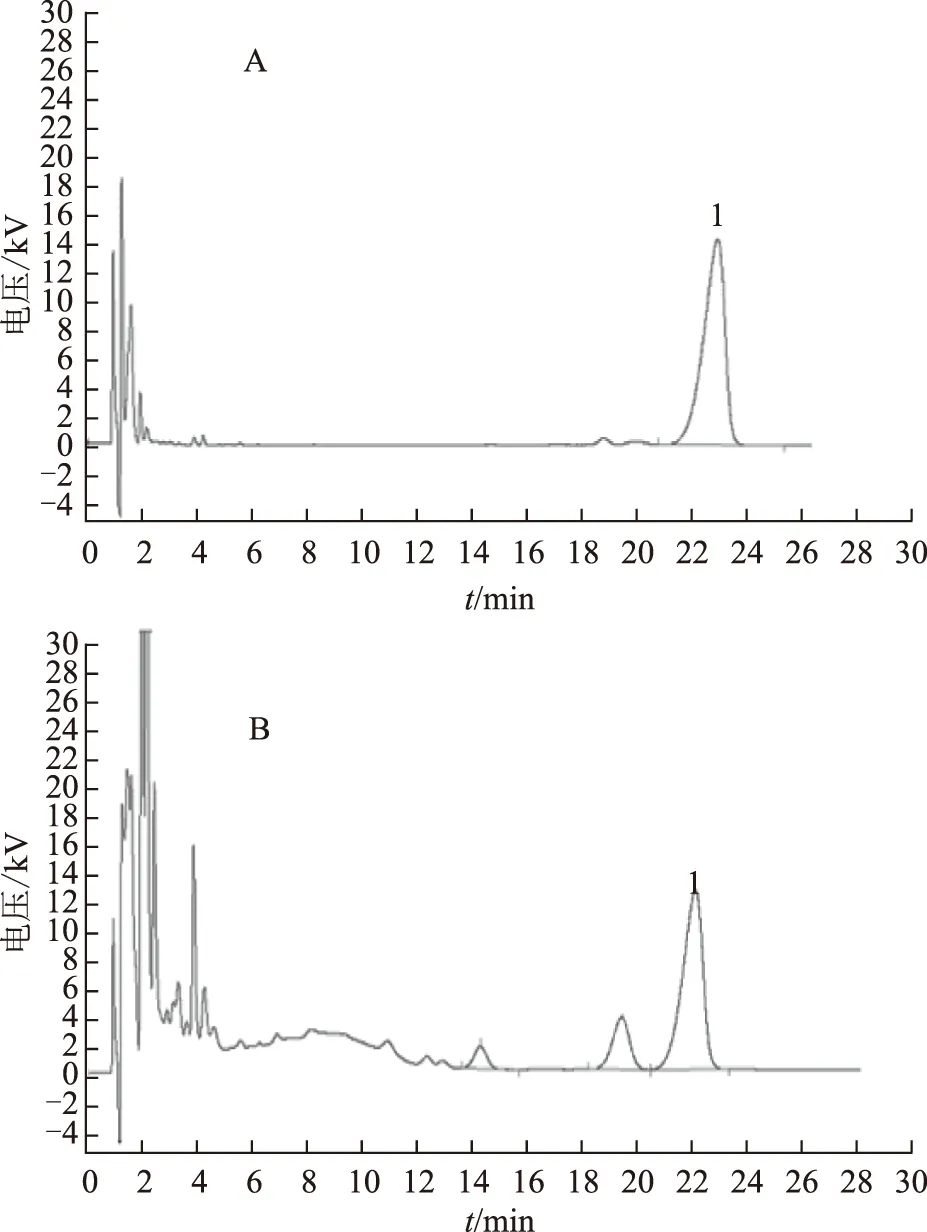
注:A.對照品;B供試品;1.人參皂苷Rb3。圖2 對照品和供試品液相色譜圖
2.3工藝條件及參數(shù)的優(yōu)化
2.3.1大孔樹脂預(yù)處理 新的D101大孔樹脂在使用前,用5倍量95%乙醇浸泡24h,用水洗至無醇味,繼用5%HCl浸泡2~3h,用水沖洗至中性,再用5%NaOH浸泡2~3h,用水沖洗至中性,備用。
2.3.2上柱溶液的制備 稱取西洋參葉適量,用12倍量水煎煮2次,每次1h,濾過,合并濾液,定容至一定體積,即得上柱溶液(每mL含0.04g生藥),備用。經(jīng)測定總皂苷含量為4.640mg·mL-1;人參皂苷Rb3含量為1.073mg·mL-1。
2.3.3大孔吸附樹脂對西洋參葉總皂苷靜態(tài)飽和吸附量與解吸率的測定 靜態(tài)飽和吸附量的測定[7]:準(zhǔn)確稱取已處理好的D101大孔吸附樹脂15.0g,置于250mL錐形瓶中,準(zhǔn)確加入西洋參葉上柱溶液200mL,在100r·min-1的條件下振蕩24h,使之充分吸附,濾過,取續(xù)濾液10mL,蒸干,照2.1.2項下方法自“殘渣加水10mL使溶解”起,依法制得供試品溶液,并測定吸附剩余液中總皂苷及人參皂苷Rb3含量,計算D101大孔樹脂在室溫條件下對西洋參葉莖葉總皂苷及人參皂苷Rb3的飽和吸附量。
吸附量=[(吸附前濃度-吸附后濃度)×溶液體積]/樹脂質(zhì)量,測定結(jié)果見表1。
靜態(tài)解吸實驗:經(jīng)過24h靜態(tài)吸附的樹脂淋凈液體,準(zhǔn)確加入200mL80%的乙醇,室溫下連續(xù)振蕩12h,使皂苷充分解吸附,濾過,取續(xù)濾液,測定總皂苷及人參皂苷Rb3含量,計算解吸率。
解吸率(%)=[(解吸后溶液濃度×解吸后溶液體積)/(吸附前溶液濃度×吸附液體積-吸附后溶液濃度×吸附液體積)]×100%,測定結(jié)果見表1。

表1 靜態(tài)飽和吸附量與解吸率的測定(n=3)
2.3.4 大孔吸附樹脂對西洋參葉總皂苷動態(tài)飽和吸附量與解吸率的測定 動態(tài)飽和吸附量的測定:取西洋參葉上柱溶液,緩慢通過裝有D101大孔樹脂15 g的柱,控制流速為1.5 mL·min-1,每10.0 mL為1個流份,收集20份,蒸干,照2.1.2項下方法自“殘渣加水10 mL使溶解”起,依法制得供試品溶液,并測定各流份中總皂苷及人參皂苷Rb3的含量。以流出液中總皂苷的含量為縱坐標(biāo),流出液收集管數(shù)為橫坐標(biāo)繪制動態(tài)吸附流出曲線,吸附流出曲線見圖3。
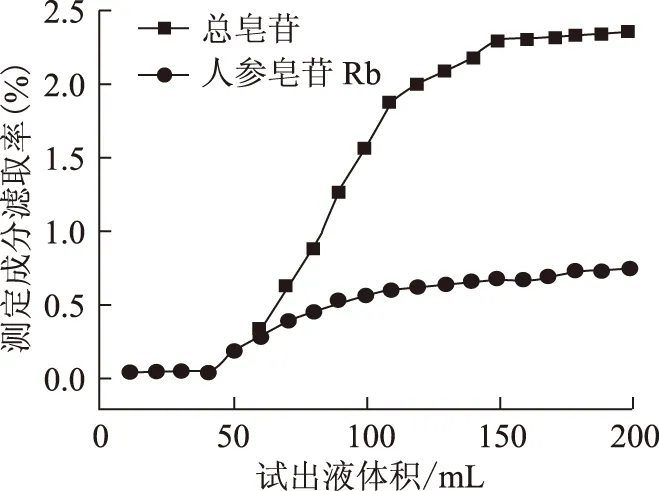
圖3 西洋參葉提取溶液動態(tài)吸附流出曲線
結(jié)果表明,從第11流份開始,流出液顏色逐漸加深,皂苷出現(xiàn)逐漸泄漏現(xiàn)象,第16份時洗出液中總皂苷含量已達原藥液含量的50%,并一直持續(xù)至20份,但未出現(xiàn)洗出液中總皂苷含量與上柱溶液相當(dāng)?shù)牧鞣荨?/p>
動態(tài)解吸實驗:動態(tài)吸附飽和的樹脂,用水洗至近無色,再以80% 乙醇洗脫,收集80%乙醇洗脫液,并定容至100 mL,精密吸取5 mL,蒸干,照2.1.2項下方法自“殘渣加水10 mL使溶解”起,依法制得供試品溶液,并測定總皂苷及人參皂苷Rb3含量,計算D101大孔樹脂在室溫條件下對西洋參葉莖葉總皂苷及人參皂苷Rb3的飽和吸附量,測定結(jié)果見表2。

表2 動態(tài)飽和吸附量和解吸率的測定(n=3)
結(jié)果提示,上柱溶液反復(fù)多次通過D101吸附樹脂,可促使吸附樹脂接近飽合。為避免生產(chǎn)過程中造成總皂苷的泄漏損失,結(jié)合靜態(tài)吸附實驗結(jié)果,以樹脂吸附量的80%上樣,即每g大孔樹脂總皂苷的上樣量不得超過34 mg;人參皂苷Rb3的上樣量不超過6.5 mg,即10 kg樹脂上樣量不超過3 kg西洋參葉藥材。
2.3.5 洗脫溶媒的選擇 采用乙醇-水系統(tǒng)為洗脫液,取經(jīng)過24 h靜態(tài)吸附的樹脂,先用3倍柱體積的水洗脫,再依次用4倍柱體積的10%、20%、30%、50%、70%、80%的乙醇連續(xù)洗脫,各洗脫液分別定容至100 mL,取各洗脫液5~20 mL,制備供試品溶液,照含量測定方法分別測定總皂苷和人參皂苷Rb3的含量,計算洗脫率,洗脫曲線見圖4。
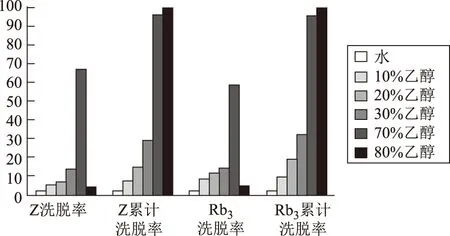
圖4 不同濃度乙醇洗脫曲線
由圖4可知,用70%乙醇溶液洗脫總皂苷及人參皂苷Rb3,洗脫量均最高,且累計洗脫率均在95%以上,故選擇70%乙醇溶液作為洗脫溶媒。
2.3.6 洗脫溶媒用量 取經(jīng)過24 h靜態(tài)吸附的樹脂,先用3倍柱體積的水洗柱除,去水溶性雜質(zhì),然后用70%乙醇溶液洗脫,分段收集,定容,測定洗脫液中總皂苷的含量及人參皂苷Rb3含量,以洗脫液中人參皂苷的含量為縱坐標(biāo),收集管數(shù)為橫坐標(biāo)繪制動態(tài)脫附曲線。計算洗脫率,確定洗脫溶媒用量。洗脫溶溶媒用量曲線見圖5。
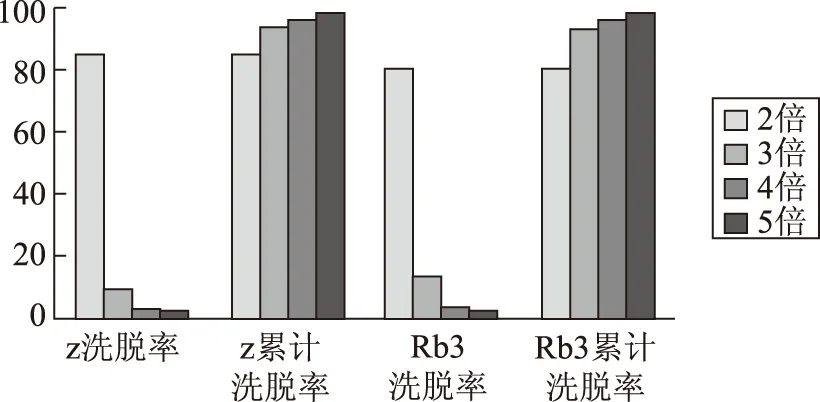
圖5 洗脫溶媒用量曲線
由圖5可知,三倍體積70%乙醇可使總皂苷及人參皂苷Rb3的洗脫率在90%以上,由此確定解吸洗脫溶液的用量為柱體積的3倍。
2.3.6 上柱溶液濃度和上樣流速試驗 因素水平:以上柱液濃度(A)、吸附流速(B)為因素,總皂苷含量及提取物得率為考察指標(biāo)進行全面試驗。因素水平見表3。
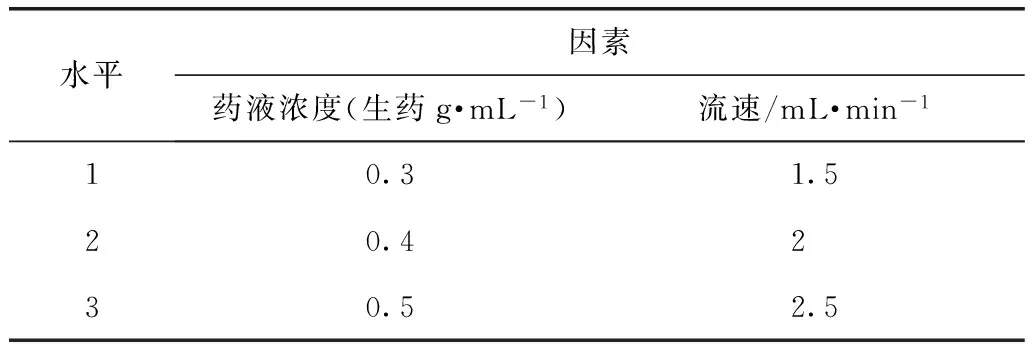
表3 上柱溶液濃度和上樣流速試驗水平表(n=3)
全面試驗:稱取西洋參葉60 g,平行9份,用12倍量水煎煮2次,每次1 h,濾過,合并濾液,濃縮并定容至一定體積,制得不同濃度的上柱溶液。按表3條件,通過吸附樹脂(裝量200 g)柱,依次分別用3倍體積的水、3倍體積70%乙醇洗脫,收集70%乙醇洗脫液,減壓、濃縮、干燥,即得總皂苷提取物。采用2.1項下方法,測定總皂苷含量,結(jié)果見表4。
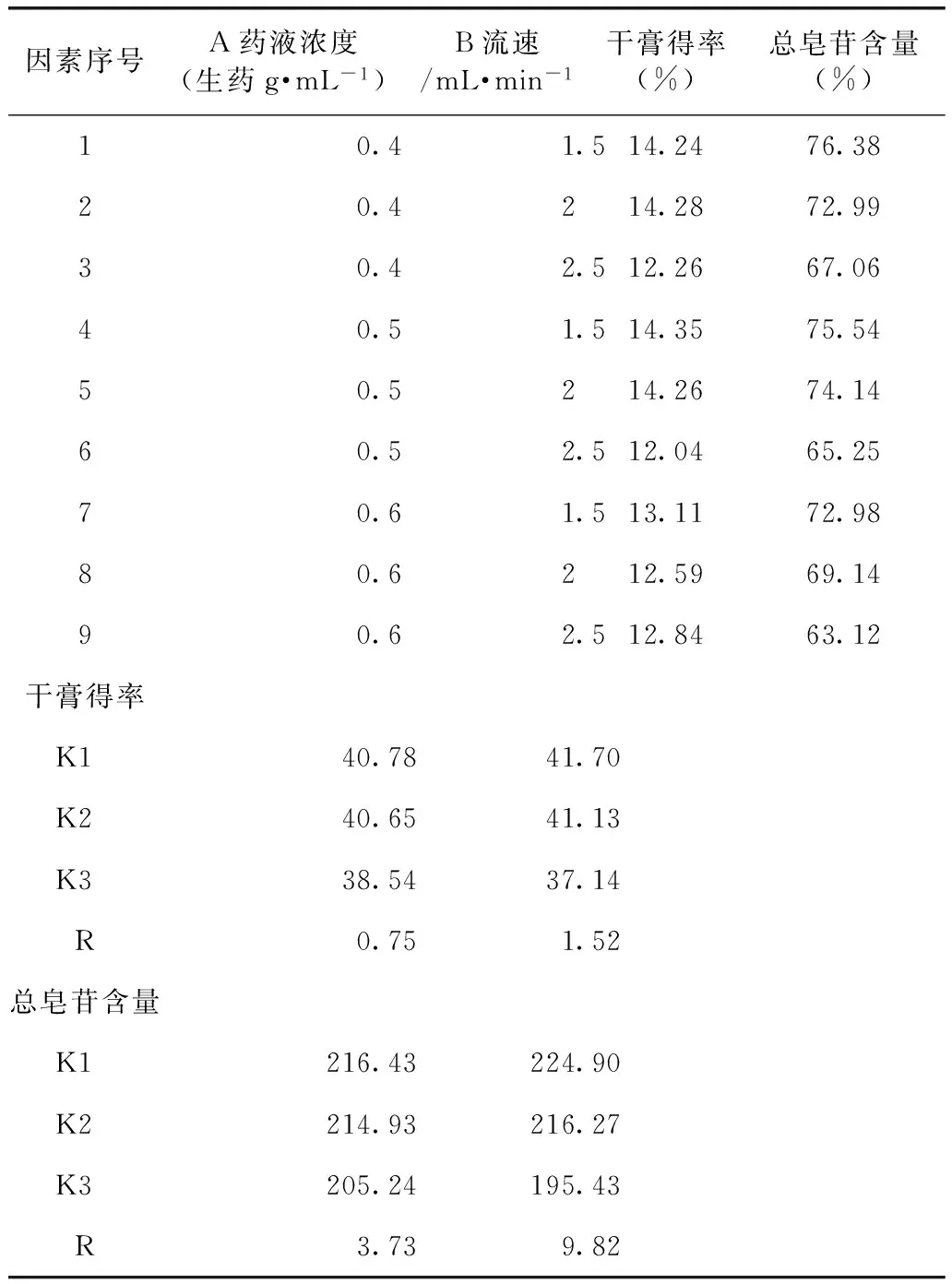
表4 上柱溶液濃度和上樣流速的試驗結(jié)果(n=9)
從表4可以看出,隨著上樣液濃度及吸附流速的增加,干膏得量及干膏中總皂苷含量有所變化;為保證提取效率及提取效果,選用上柱溶液濃度為0.4 g·mL-1,上樣流速為1.5 mL·min-1。
2.3.7 重復(fù)驗證試驗 稱取西洋參葉3 kg,加12倍量水煎煮2次,每次1 h,濾過,濾液濃縮至6000 mL,通過裝有10 kg預(yù)處理好的D101樹脂柱,Φ200 mm×600 mm,按1.5 mL·min-1流速上樣,將流出液重復(fù)通過樹脂柱,先用蒸餾水(3 BV)洗滌,再用70%的乙醇(3 BV)洗脫,收集70%乙醇洗脫液,回收乙醇,濃縮,減壓干燥,測定干膏中總皂苷和人參皂苷Rb3的含量。結(jié)果見表5。
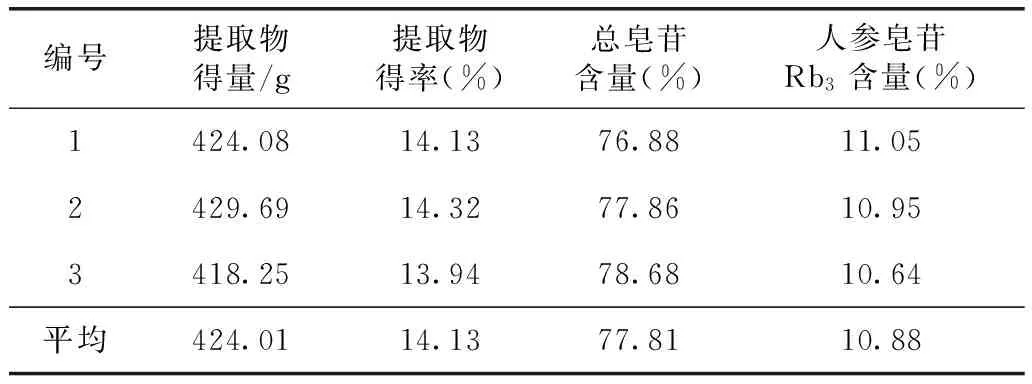
表5 重復(fù)驗證結(jié)果(n=3)
3 討論
驗證試驗表明,西洋參葉加12倍量煎煮提取2次,每次1h,濃縮至每ML含0.5g生藥,以D101大孔樹脂進行吸附,使用濃度70%的乙醇進行洗脫,即可獲得高收率、高含量的皂苷提取物,總皂苷含量大于75%,該方法操作簡便且易于實現(xiàn)工業(yè)化。
本研究中,用分光光度法與HPLC法結(jié)合的雙指標(biāo)測定方法來考察工藝條件和參數(shù),而在考察上柱溶液濃度和上樣流速時僅采用了分光光度法,為的是縮短分析時間,同時達到含量測定結(jié)果準(zhǔn)確。
在試驗中表明,西洋參葉90℃溫浸2h,與煎煮提取1.0h的效果相當(dāng)。
棄去水及30%乙醇洗脫液,收集70%洗脫液,所得提取物中總皂苷含量沒有明顯變化,而人參皂苷Rb3含量明顯提高。
將上柱溶液重復(fù)通過樹脂柱,可提高干膏得率及提取物中總皂苷和人參皂苷Rb3的含量,但在動態(tài)吸附考察時未找到吸附飽和點,初步分析可能是吸附樹脂的吸附性與時間有關(guān),其內(nèi)在聯(lián)系有待進一步研究。
[1] 魏曉雨,田義新,趙智靈,等.我國西洋參引種30年后遺傳穩(wěn)定性研究[J].中國中藥雜志,2014,39(19):3723-3726.
[2] 阮征,姜永濤,李國強.西洋參葉中皂苷類成分指紋圖譜研究[J].中國現(xiàn)代中藥,2011,13(3):3723-3726.
[3] 辛艷茹,馬萍,張英娜.西洋參葉的研究進展[J].時珍國醫(yī)國藥,2001,12(8):274.
[4] 楊瑩瑩,張廣晶,張舒媛.大孔樹脂在中藥研究中的應(yīng)用[J].遼寧中醫(yī)雜志,2014,41(10):2193-2195.
[5] 國家藥典委員會.中華人民共和國藥典:一部[S].北京:中國醫(yī)藥科技出版社,2010.
[6] 王隸書,李陽,程東巖.HPLC法測定通栓凍干粉針中人參皂苷Rb3的含量[J].中國實驗方劑學(xué)雜志,2005,11(3):11-12.
[7] 辛海量,盛佳鈺,葛凌弦,等.人參葉提取物的制備工藝研究[J].醫(yī)藥導(dǎo)報,2011,30(1):79-82.
PurificationofTotalSaponinsofLeavesofPanaxquinquefoliusbyMacroreticularResin
GUOTingting1,WANGZhaohua2*,ZHANGDajun2
(1.TheAffiliatedHospitalToChangchunUniversityofChineseMedicine,Changchun130021,China;2.AcademyofChineseMedicalSciencesofJilinProvince,Changchun130012,China)
Objective:The study aims at purification of total saponins of leaves ofPanaxquinquefloiusby D-101macroreticular resin.Methods:Using the adsorption and desorption rates of total saponins and ginsenside Rb3as parameters,the purification technological conditions of total saponins of the leaves ofP.quinquefloiusby macroreticular resin were experimentally investigated.The curves of adsorption kinetics and dynamic adsorption kinetics of macroreticular resin were exploited.Results:The rate of medicinal materials and the wet resin content was3∶10.The flow rate of the macro-reticular resin column was1.5mL·min-1.The experiments were repeated twice.The column was eluted by water of three times volume and ethanol of three times volume.After then ethanol elution was collected.The ethanol elution was concentrated,dried,and then the total saponins extract of the leaves ofP.quinquefloiuswas obtained.The contents of total saponins in extract was more than75%.The contents of ginsenside Rb3was more than10%.The yield of total saponins extract was14%.Conclusion:This method is simple,repeatable,available for extract and purification of total saponins of the leaves ofP.quinquefloius.
Leaves ofPanaxquinquefloius;macroreticular resin;total saponins;ginsenside Rb3;technological conditions and parameters
10.13313/j.issn.1673-4890.2016.9.025
2015-05-04)
國家科技重大專項(2013ZX09103002-022);吉林省科技發(fā)展計劃專項(20140311014YY)
*
王兆華,主任藥師,研究方向:中藥新藥與新制劑;Tel:(0431)86058659,E-mail:wangzhaohua221@163.com
