銀黃清肺膠囊化學成分的LC-ESI-MS/MS分析
2016-10-17 08:53:48吳志軍蔣學華
中國測試 2016年3期
關鍵詞:檢測
汪 丹,蔡 甜,吳志軍,蔣學華
(1.四川大學華西藥學院,四川成都610041;2.成都大學醫學院,四川成都610106;3.中國科學院成都生物所,四川成都610041)
銀黃清肺膠囊化學成分的LC-ESI-MS/MS分析
汪丹1,2,蔡甜3,吳志軍3,蔣學華1
(1.四川大學華西藥學院,四川成都610041;2.成都大學醫學院,四川成都610106;3.中國科學院成都生物所,四川成都610041)
研究液質聯用技術對銀黃清肺膠囊化學成分的分析。對銀黃清肺膠囊提取物進行LC-MS分析,確定分子式,鑒定出部分質譜峰的結構。共發現36種化合物,其中有蔗糖、尿苷、2,3-二脫氧尿苷、沒食子酸、奎尼酸、新綠原酸、表沒食子兒茶素、原兒茶酸、莽草酸、阿魏酸、咖啡酸、反式阿魏酸、犬尿喹啉酸、羥基苯甲酸、4-甲氧基芥子酸、山奈酚、異鼠李素、大黃素18種未在正離子模式下的液質聯用文獻中報道過。復方中藥的有效成分LC-MS檢測,正離子模式適用于檢測大多數雜原子化合物,負離子模式更適用于含羧基、多羥基的化合物(如酚酸、多酚類、部分黃酮和苷類)的檢測,為銀黃清肺膠囊進一步質量研究及檢測方法選擇提供理化依據。
銀黃清肺膠囊;LC-ESI-MS;酚酸;黃酮
0 引 言
銀黃清肺膠囊是由14味中藥制成的用于清肺化痰、止咳平喘的復方中藥制劑,其組成藥材有北葶藶子、麻黃(炙)、苦杏仁、浙貝母、枇杷葉、大青葉、石菖蒲、穿山龍、一只蒿、銀杏葉、五味子、枳實、生石膏、甘草[1]。它的名稱易與銀黃制劑[2-3](由金銀花和黃芩提取物制成)混淆,兩者藥理作用不同,后者主治急性扁桃體炎及上呼吸道感染。現有研究中,周卿意駿等[4]使用HPLC-Q-TOF-MS/MS的技術,在正離子模式下鑒定出了其中54個成分,并嘗試建立了10批次藥材中14個成分的指紋圖譜[1],但是活性成分中的含羧酸、酚酸基團的組分及部分黃酮、苷類等在正離子模式下響應較弱,不易被檢測,更適合在負離子模式下進行測定。
1 實驗部分
1.1儀器與試劑
Agilent 1100型高效液相色譜儀配G1315B-DAD檢測器(美國Agilent科技有限公司);micrOTOF-Q質譜儀(德國Bruker公司);AUX120型萬分之一電子天平(日本島津公司);AFZ-1001-U型艾科浦超純水系統(美國艾科浦國際有限公司)。
銀黃清肺膠囊樣品批號131003(湖南安邦制藥有限公司的獨家品種);甲醇為色譜級(Fisher ScientificPittsburgh,PA,USA);綠原酸、阿魏酸、對香豆酸、槲皮素、蘆丁、異鼠李素、大黃素、表沒食子兒茶素、兒茶素、咖啡酸、沒食子酸標準品購于四川省維克奇生物科技有限公司,含量>98%。山奈酚、3,4-二羥基苯甲酸(原兒茶酸)標準品購于成都瑞芬思生物科技有限公司,含量>98%。
1.2實驗方法
1.2.1樣品及空白對照液的制備
樣品溶液:取銀黃清肺膠囊,抖出內容物,精密稱定0.15g,置于1.5mL帶蓋的玻璃試劑瓶中,精密量取甲醇600 μL,密封浸漬2 h后,超聲15 min,吸取試液,于12000r/min離心10min,取上清液進樣。
空白對照液:按上述方法,不加藥物,制備空白對照液。
1.2.2分析條件
液相色譜條件:色譜柱:Inertsil ODS-4(250mm× 4.6mm×5μm,GL sciences Inc);流動相為0.1%甲酸水溶液(A)和甲醇(B),梯度洗脫,洗脫程序如下:0~2 min,10%B;2~5 min,10%~50%B;5~15 min,50%~100%B;15~25 min,100%B;柱溫30℃,進樣量15 μL,流量0.8 mL/min,用三通管分流后連接質譜進樣。
質譜條件:離子源為ESI源;在負離子條件下檢測,End Plate Offset-500V;Capillary,3 500V;氦氣作為碰撞氣體,高純度氮氣作為霧化和干燥氣體,流量為6L/min,壓力為1.0bar(1bar=105Pa),干燥氣溫度180℃。數據采集范圍50~1500m/z。質量數據使用Bruker數據分析4.0版軟件處理。預試后,選擇12eV作為MS2的主要碰撞能,對于12eV打不碎的山奈酚、大黃素,設為20eV。
2 結果與討論
采用LC-MS技術,可獲得各質譜峰的MS信息,利用Bruker數據分析4.0版軟件,可獲得化合物的可能分子式信息,再依據化合物的MS2碎片信息進行交叉對比,最終可確定該質譜峰的分子式。同時通過與對照品的保留時間和同條件下的碎片信息比較,或與ESI離子源的其他文獻信息進行對比后,最終可鑒定部分化合物的結構。
2.1與其他檢測方法比較的優勢
由圖1可知,LC-MS的離子流圖雖然也和HPLC的圖類似,但是各化合物不必完全分離開,只要化合物的分子量不同即使同時出峰,也不影響采集和辨識,故采集時間大大的縮短。而Q-TOF MS比QQQ MS的分辨率更高,可以采集到小數點后四位,避免分子量相近的離子峰相互混淆,故適用于鑒定復雜混合物的化學組分的結構。
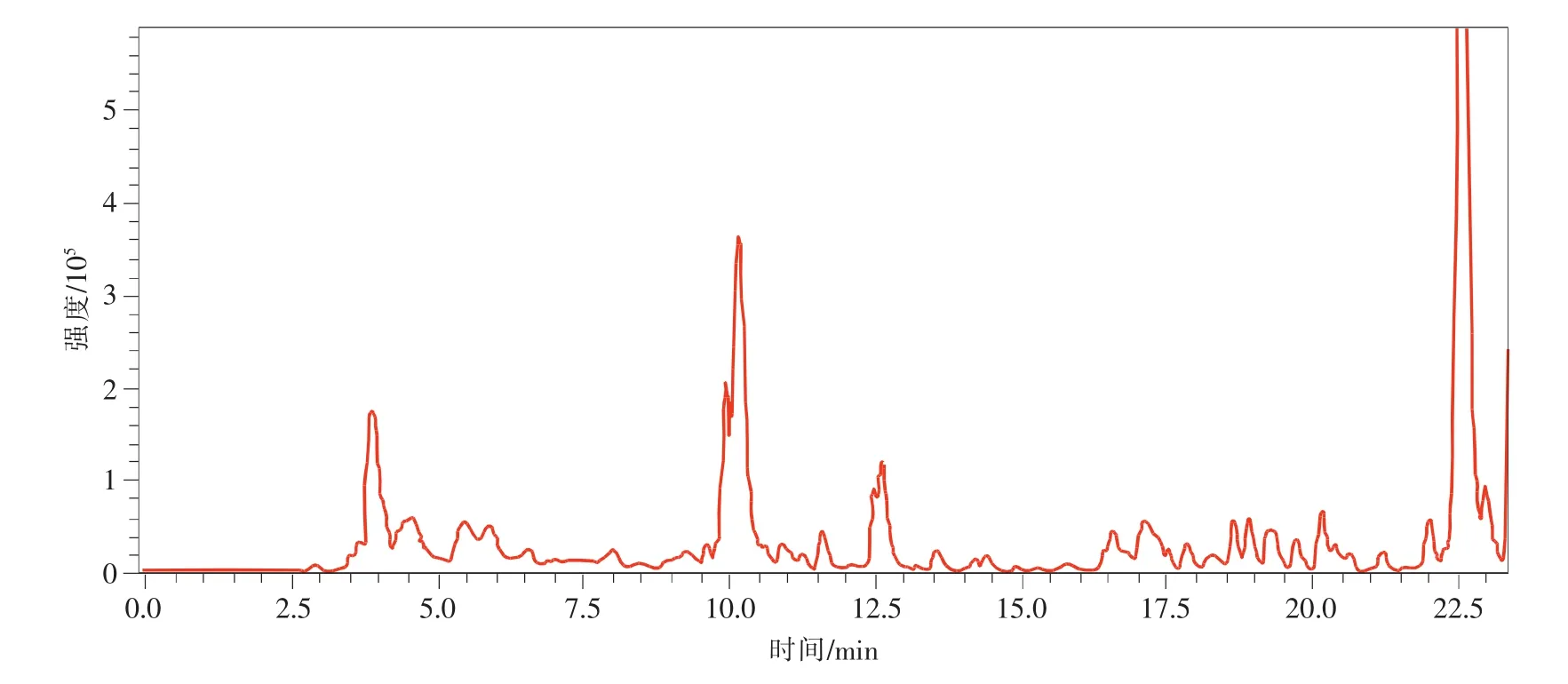
圖1 銀黃清肺膠囊的Base Peak Chromatogram離子流圖
2.2負離子模式下的檢測結果與討論
實驗共檢測到銀黃清肺膠囊提取物在負離子條件下的133組MS2碎片信息,篩除空白背景峰、裂解碎片再次碎裂后的信息,對比對照品或文獻,共鑒定出26個化合物(質譜信息見表1)。其中,蔗糖、尿苷、2,3-二脫氧尿苷、沒食子酸、奎尼酸、新綠原酸、表沒食子兒茶素、原兒茶酸、莽草酸、阿魏酸、咖啡酸、反式阿魏酸、犬尿喹啉酸、羥基苯甲酸、4-甲氧基芥子酸、山奈酚、異鼠李素、大黃素18種化合物在正離子模式下的LC-MS[4]中未被報道。
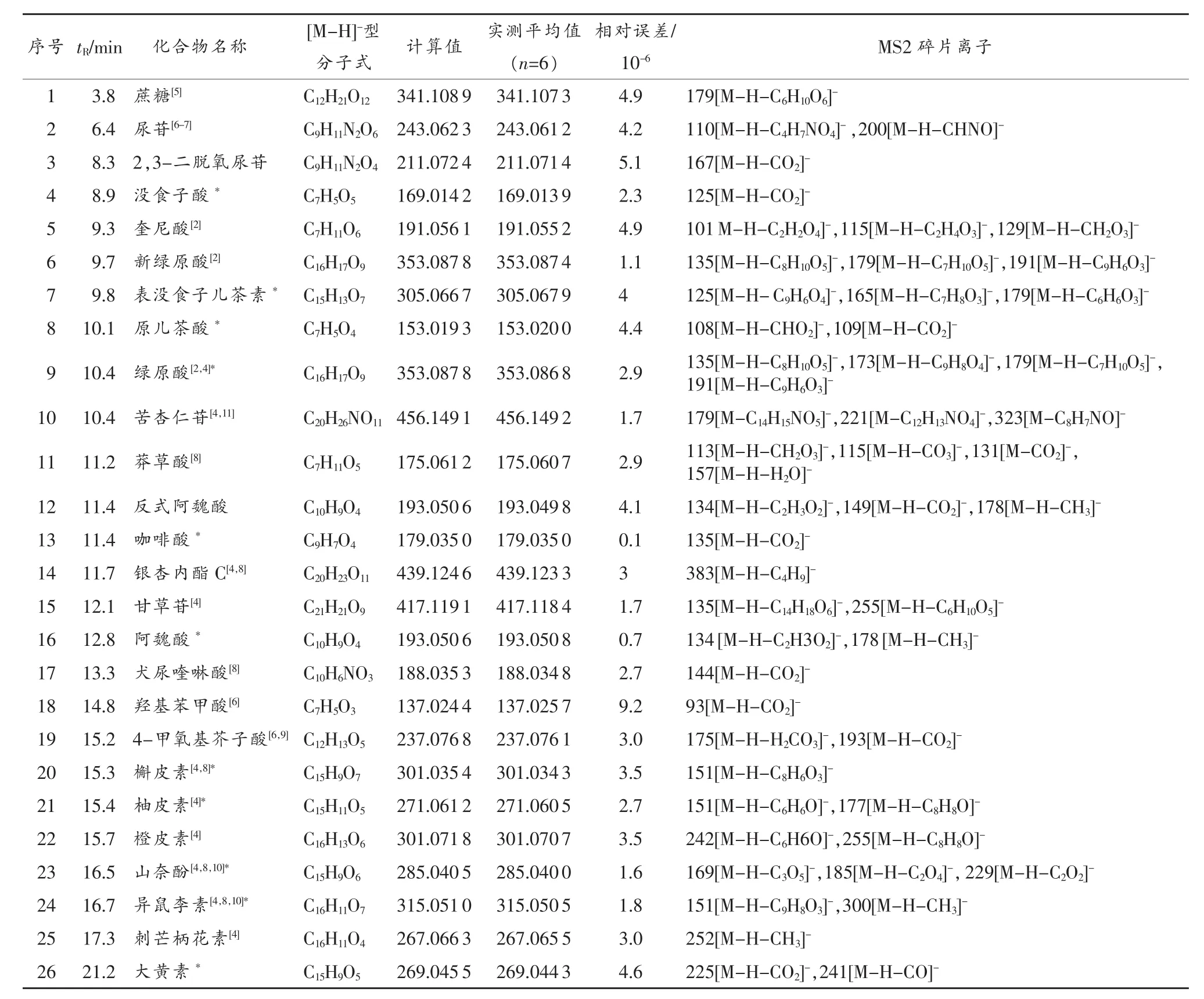
表1 化合物1~26質譜信息1)
糖類:
化合物1,分子式為C12H22O12,依據其有179的碎片以及C6H10O6的中性丟失,推測結構為兩個六碳糖相連,根據文獻[5]推斷為蔗糖。
核苷類:
化合物2,依據碎片信息和N規則,推測分子式中應含N元素,故推測其分子式為C9H12N2O6,結合中性丟失和文獻[6-7]后確定為尿苷(尿嘧啶核苷)。同理化合物3的分子式為C9H12N2O4,結合碎片及中性丟失,推斷為2,3-二脫氧尿苷。
酸類:
化合物4分子式為C7H6O5,經與沒食子酸標準品對比,保留時間、MS2碎片均一致,確定為沒食子酸。同理,與標準品對比保留時間、MS2碎片、中性丟失發現:化合物8與原兒茶酸,化合物12與阿魏酸,化合物13與咖啡酸均一致,分別鑒定化合物8、化合物12、化合物13為原兒茶酸、阿魏酸和咖啡酸。化合物16與化合物12的碎片相似,鑒定為反式阿魏酸。
化合物5、6、9,其分子式分別為C7H12O6,C16H18O9,C16H18O9。其中化合物6,9碎片峰相似,為同分異構體,經與綠原酸標準品對比后發現,化合物9為綠原酸,依據文獻[2]報道結合保留時間,化合物6為新綠原酸。而化合物5的碎片信息與綠原酸中191碎片的MS3一致,故為奎尼酸。
化合物7的分子式為C15H14O7。在按[M-H]-分子量m/z 305.0667+0.01提取離子流時發現,得到3個質荷比相似的物質峰,其保留時間分別為9.2,9.8,10.5min。進一步分析各自的碎片結構發現,保留時間為10.5min的化合物含有硫元素,分子式不一致,保留時間9.2min的化合物碎片與標準品不符,只有保留時間為9.8min的MS2碎片與表中沒食子兒茶素標準品一致。故鑒定保留時間9.8min的峰為表中沒食子兒茶素,其余兩個峰因無標準品或相關文獻未鑒定出結構。
化合物11、18、19的分子式分別為C7H12O5、C7H6O3、C12H14O5,由不飽和度推斷,三者均具有苯環結構。由碎片推斷,化合物11含有羧基和鄰羥基結構,再依據文獻[8],故推斷為莽草酸。化合物18,依據其有CO2的中性丟失,推測具有羧基,同時具有羥基,根據文獻報道其含有對羥基苯甲酸和間羥基苯甲酸,與文獻報道也是一致的。故化合物18應為羥基苯甲酸,但其所含羥基是在對位還是間位,由于未與標準品對比暫無法確定。同理,化合物19與化合物18同樣具有羧基和苯環結構,與文獻[6,9]對比后推斷為4-甲氧基芥子酸
黃酮及黃酮苷類:
依據化合物的分子式、分子量、碎片特征和保留時間與標準品對比,鑒定化合物20、23、24、26分別為槲皮素、山奈酚、異鼠李素、大黃素[4,8,10]。與文獻[4]對比MS,MS2信息,鑒定化合物15、21、22、25分別為甘草苷、柚皮素、橙皮素、刺芒柄花素。
需要注意的是,化合物23及山奈酚標準品在LC-MS實驗中的碎片相同,但與山奈酚標準品直接質譜進樣所得的碎片有不同,多出了m/z 223,239,241等峰度較大的碎片峰。進一步觀察圖譜發現,LC-MS實驗中的化合物23和LC-MS實驗中山奈酚標準品的m/z 285.0390的峰附近同時還有一個m/z 285.17的背景峰(出峰時間15~18min,碎片 223,239,241)。由于儀器在選擇碰撞時,最小的選擇m/z范圍為X+0.5,在同時出峰的情況下無法區分開 m/z 285.039和m/z 285.17,故帶來了碎片干擾。調整實驗條件,將碰撞能量增大為20eV后,山奈酚的碎片峰豐度增加,再篩除掉285.17的碎片峰m/z 223,239, 241后,化合物23可觀察到與標準品山奈酚直接質譜進樣相同的碎片,進一步確定化合物23為山奈酚。其他同樣需要20eV碰撞能才能較好碎裂的物質還有化合物26大黃素。
其他:
化合物10的分子式為C20H27NO11,其碎片與文獻[4,11]中苦杏仁苷的報道一致,故確定為苦杏仁苷。
此外檢測到分子式為C11H12O5的化合物,但實驗得到碎片結構和文獻中提到的葶藶子的化學成分已報道[4,9]的芥子酸有差異,未鑒定出結構。除此之外還檢測到多組含硫元素或磺酸基[4,12]的化合物,但其具體結構還需要進一步研究確定。
2.3與正離子模式下文獻的檢測結果對比
考慮到負離子下不易出峰的化合物,在負離子模式下的Q-TOF-MS的檢測限相對較高,有可能某些檢測到的物質峰由于峰強較低,無法得到MS2碎片,影響鑒定結構。為方便日后建立銀黃清肺膠囊中更多峰的指紋圖譜,故對銀黃清肺膠囊正離子模式下已報道的化合物,在負離子模式下,進行相應的離子流提取(m/z X+0.01%),通過其精確分子量、Err(10-6)、mSigma值(同位數比值)確定分子式,結合文獻對比得到以下10種化合物[4],如表2所示。
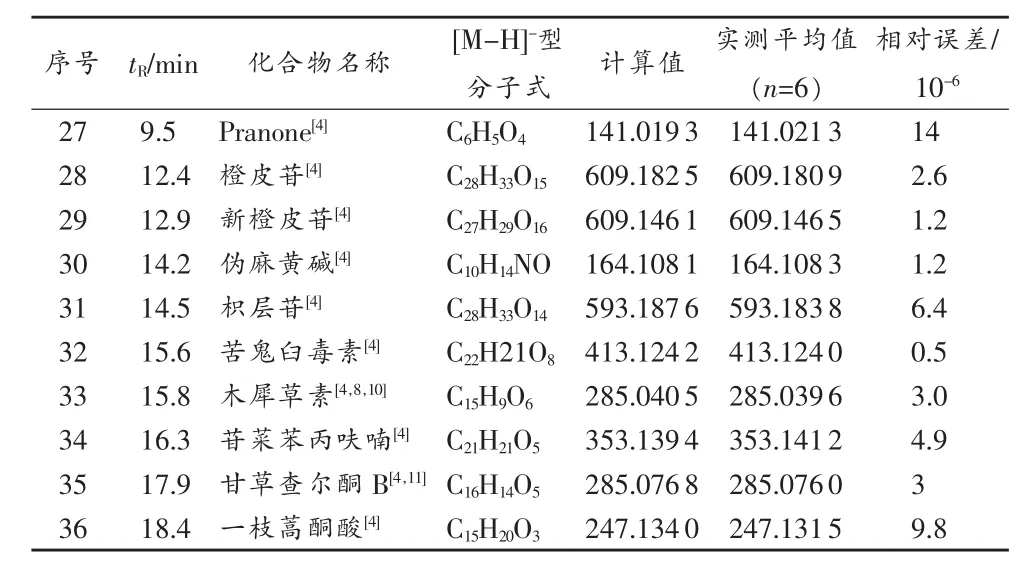
表2 化合物28~37 HPLC-ESI-Q-TOF信息
表中除化合物27、31和36以外,其余化合物的誤差均小于5×10-6,符合準確確定分子式的要求。
分析化合物27、化合物31、化合物36誤差偏大的原因:在濃度接近檢測限時,化合物的準確質量采集可能出現相對較大的偏差。化合物27推斷為文獻[4]中所報道的Pranone(分子式為C6H6O4),其實測值與理論值m/z相差0.002 0,低于Q-TOF儀器m/z X+0.003的系統誤差,只不過由于其理論分子量較小(141.0193),算出來的相對誤差(10-6)偏大。同理,化合物31、36推斷為文獻[4]中所報道的枳層苷、一枝蒿酮酸,其相對誤差均小于10×10-6。
據文獻報道,樣品中可能含[M-H]-的m/z 609的黃酮苷化合物有橙皮苷、新橙皮苷和蘆丁3種。化合物28依據其準確分子量和分子式推斷為文獻[4]中所報道的橙皮苷。化合物29有新橙皮苷和蘆丁兩種可能,且新橙皮苷和蘆丁為同分異構體,分子式相同。進一步用蘆丁標準品進行實驗比較,蘆丁的保留時間為12.6 min,與化合物29不一致,故推斷化合物29為新橙皮苷。
化合物30依據測得的分子式可能為麻黃或偽麻黃堿這一對異構體中的一種,但是比較文獻中的保留時間,因三者的極性差異,在反相柱中的出峰順序為麻黃堿先于Pranone,偽麻黃堿最后出峰,故推斷化合物30為偽麻黃堿。
化合物32~35由5×10-6以內得到的分子式與文獻對比推斷為苦鬼臼毒素、木犀草素、苷菜苯丙呋喃、甘草查爾酮B[4,8,10-11]。
由上述結果可知,大部分正離子模式的文獻中檢測到的含雜原子的離子峰在負離子模式下響應較弱,不利于檢測。而負離子模式下也采集到18種在正離子模式下不易出峰的酚類或黃酮類化合物。正負離子模式下物質峰有較明顯差異。
3 結束語
本研究建立了銀黃清肺膠囊中復雜成分的簡易、快速的LC-MSn分析方法,能夠同時實現多組分的良好分離并提供各峰的組成和結構信息,共找到了36種化合物,其中有蔗糖、尿苷、2,3-二脫氧尿苷、沒食子酸、奎尼酸、新綠原酸、表沒食子兒茶素、原兒茶酸、莽草酸、阿魏酸、咖啡酸、反式阿魏酸、犬尿喹啉酸、羥基苯甲酸、4-甲氧基芥子酸、山奈酚、異鼠李素、大黃素18種未在正離子模式下的文獻中報道。由實驗和文獻對比可知,正負離子模式下出峰的物質差異較大,復方中藥的有效成分LC-MS檢測,正離子模式適用于檢測大多數含雜原子的化合物,而負離子模式更適用于含羧基、多羥基的化合物(如酚酸、多酚類、部分黃酮和苷類)的檢測,應同時關注兩種測試條件下的結果才完整。本文為銀黃清肺膠囊的質量研究及檢測方法的合理選擇奠定了基礎。
[1]周卿意駿,張水寒,高尚,等.銀黃清肺膠囊HPLC指紋圖譜研究[J].中草藥,2015,46(9):1314-1320.
[2]趙剛,杜瑋,魏玉輝,等.銀黃制劑的HPLC-ELSD指紋圖譜研究[J].中草藥,2009,40(7):1053-1056.
[3]向青,王小花,林慧,等.HPLC-DAD-Q-TOF-MS/MS法的銀黃顆粒主要成分定性與定量研究[J].中成藥,2015,37(1):105-112.
[4]周卿意駿.銀黃清肺膠囊指紋圖譜的研究[D].湖南:湖南中醫藥大學,2015.
[5]榮立新.石菖蒲化學成分的研究進展 [J].中國民間療法,1995(6):44.
[6]周喜丹,唐力英,周國洪,等.南北葶藶子的最新研究進展[J].中國中藥雜志,2014,39(24):4699-4708.
[7]毛豐,金藝,袁波,等.尿苷和表告依春在大鼠體內的藥物動力學[J].沈陽藥科大學學報,2014,31(4):301-308.
[8]陳晶.銀杏葉_銀杏葉提取物及其注射液中化學成分及酚酸含量的研究[D].北京:北京中醫藥大學,2013.
[9]CHENG B F,HOU Y Y,WANG L Q,et al.Dualbioactivity-basedliquidchromatography-coupled quadrupole time-of-flight mass spectrometry for NF-κB inhibitors and β2AR agonists identification in Chinese Medicinal Preparation Qingfei Xiaoyan Wan[J].Anal Bioanal Chem,2012(404):2445-2452.
[10]柳文媛,周晨,閆翠敏,等.HPLC-DAD-ESI-MS/MS法用于枳實提取物的化學成分分析及多組分同時定量測定[J].中國天然藥物,2012,10(6):456-463.
[11]李睿,曾岑,王平,等.基于GC-MS和UPLC-Q-TOF-MS的麻黃湯化學成分識別[J].中國中藥雜志,2014,39(4):704-709.
[12]薛坤,劉雅琴.石膏配伍應用對復方化學成分的影響[J].天津藥學,2014,26(5):47-50.
(編輯:徐柳)
LC-ESI-MS/MS analysis of chemical constituents in Yinhuang Qingfei capsule
WANG Dan1,2,CAI Tian3,WU Zhijun3,JIANG Xuehua1
(1.West China School of Pharmacy,Sichuan University,Chengdu 610041,China;2.School of Medicine and Nursing,Chengdu University,Chengdu 610106,China;3.Chengdu Institute of Biology,Chinese Academy of Sciences,Chengdu 610041,China)
To analyze chemical constituents of Yinhuang Qingfei capsules by HPLC-ESI-Q-TOFMSMS method.The extracts of Yinhuang Qingfei capsules are analyzed with LC-MS method to determine molecular formula and identify the structures of partial spectral peak.36 compounds are found,including sucrose,uridine,2,3-deoxyuridine,gallic acid,quinic acid,Neochlorogenic acid,(-)-Epigallocatechin,protocatechuic acid,shikimic acid,ferulic acid,caffeic acid,trans ferulicacid, kynurenicacid, Hydroxybenzoicacid, 4-methoxy-erucicacid, kaempferol,isorhamnetin,Emodin,18 components that are not reported in HLPC-MS literatures under the positive ion mode.In LC-MS detection of active ingredients in compound traditional Chinese medicine,the positive ion mode can be applied to detect most hetero atomic compounds while the,negative ion mode is more suitable to detect carboxyl-containing and polyhydroxy compounds(such as phenolic acid,polyphenol,flavonoid and glycoside).This paper has laid a foundation for further studying and testing the quality of Yinhuang Qingfei capsules.
Yinhuang Qingfei capsules;LC-ESI-MS;phenolic acids;flavonoids
A
1674-5124(2016)03-0036-05
10.11857/j.issn.1674-5124.2016.03.009
2015-10-18;
2015-12-08
國家自然科學基金青年項目(21305137)
汪丹(1983-),女,四川成都市人,碩士研究生,專業方向為臨床藥學及藥物MS分析。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
