利用水稻MAGIC群體關聯定位白葉枯病抗性QTL和創制抗病新種質
2016-10-19 04:13:58陳天曉朱亞軍密雪飛陳凱孟麗君左示敏徐建龍2
作物學報 2016年10期
關鍵詞:水稻
陳天曉朱亞軍密雪飛陳 凱孟麗君左示敏徐建龍2,,4,*
1揚州大學江蘇省作物遺傳生理國家重點實驗室培育點 / 糧食作物現代產業技術協同創新中心, 教育部植物功能基因組學重點實驗室, 江蘇揚州 225009;2中國農業科學院作物科學研究所, 北京 100081;3中國農業科學院深圳農業基因組研究所, 廣東深圳 518210;4中國農業科學院深圳生物育種創新研究院, 廣東深圳 518210
利用水稻MAGIC群體關聯定位白葉枯病抗性QTL和創制抗病新種質
陳天曉1,2朱亞軍3密雪飛3陳 凱3孟麗君3左示敏1,*徐建龍2,3,4,*
1揚州大學江蘇省作物遺傳生理國家重點實驗室培育點 / 糧食作物現代產業技術協同創新中心, 教育部植物功能基因組學重點實驗室, 江蘇揚州 225009;2中國農業科學院作物科學研究所, 北京 100081;3中國農業科學院深圳農業基因組研究所, 廣東深圳 518210;4中國農業科學院深圳生物育種創新研究院, 廣東深圳 518210
以 8個不同親本構建的遺傳上相互關聯的多親本高代互交系(multi-parents advanced generation inter-cross,MAGIC)群體, 包括2個4親本群體(DC1和DC2)和1個八親本群體(DC3)為材料, 接種我國白葉枯病強致病力V型菌系(GD-V)和弱致病力II型菌系(C2), 關聯分析定位MAGIC群體對白葉枯病的抗性QTL, 篩選抗病種質。結果表明, 大多數親本對C2菌系表現抗病, 而對GD-V菌系表現感病, 3個MAGIC群體的病斑長度均出現超親分離。共檢測到 7個白葉枯病抗性 QTL, 大多表現數量抗性, 而且抗性 QTL表達存在明顯的遺傳背景效應。QBbr11-1和QBbr11-2受遺傳背景影響較小, 具有一定的育種應用價值。從3個群體篩選出8份不同抗病QTL聚合的抗病材料, 表明質量抗性基因和水平抗性數量性狀位點的結合可以顯著提高抗性水平。8份不同抗病QTL的聚合系可以用作抗病育種的中間抗源。研究結果表明, MAGIC群體可以將遺傳研究和育種應用有機結合, 是遺傳研究和開展標記輔助育種的理想群體。
MAGIC; 水稻白葉枯病; QTL; 全基因組關聯分析; 水稻
由黃單胞桿菌水稻致病變種(Xanthomonas oryzae pv.oryzae, Xoo)引起的水稻白葉枯病是一種造成稻米產量損失的細菌病害。最早在日本福岡地區, 之后陸續在世界主要水稻產區發現該病, 當前已經成為我國水稻生產中的三大病害之一[1-2]。實踐證明,挖掘并利用抗性基因培育抗病品種是防治水稻白葉枯病最經濟有效的手段[3]。
白葉枯病菌與水稻抗病基因之間存在典型的特異性互作, 水稻的 R基因與病原菌無毒基因(Avr)產物之間直接或間接相互作用產生的“基因對基因”抗性, 是水稻對白葉枯病抗性表現的重要形式[4], 同時被微效多基因控制的水平抗性也在水稻白葉枯病抗性中發揮重要作用[5]。迄今, 經國際注冊確認和公開報道的水稻白葉枯病抗性基因已達30多個[6]。多數白葉枯病抗性基因由于抗譜狹窄或為隱性基因未被直接應用于抗病育種實踐, 僅 Xa3、Xa4、xa5、Xa7、xa13、Xa21、Xa23等少數抗白葉枯病基因得到較廣泛的應用[7-8], 在一定程度上降低了白葉枯病帶來的產量損失。然而, 根據“基因對基因”假說[9-10],在抗病基因的選擇壓下, 致病菌將不斷進化, 使得某些抗性主效基因逐漸失去抗性。因此, 發掘新的抗性基因培育抗病品種將是抗病育種研究永恒不變的研究方向。在白葉枯病抗病遺傳育種領域, 除直接利用抗性基因的策略外, Li等[11]也提出將垂直抗性基因和多個微效基因聚合以期獲得水稻對白葉枯病的持久抗性的策略。
起源于不同地域的種質資源中往往隱藏著豐富的有利等位基因[12]。先前, Meng等[13]以8個來自國際水稻研究所、中國和其他國家的優良種質為親本,構建了3個相互關聯的MAGIC群體, 成功開展了重要農藝性狀的QTL定位研究。在本研究中, 我們對這3個MAGIC群體接種2個致病力不同的水稻白葉枯病菌, 開展了水稻抗白葉枯病QTL的定位研究,并篩選出部分抗病中間材料。研究結果為發掘白葉枯病抗性新基因和培育抗白葉枯病水稻新品種提供了相關信息和材料基礎。
1 材料與方法
1.1 試驗群體
3 個MAGIC群體是利用來自不同育種項目的8個多樣性豐富的優良種質為親本構建的, 包括 2個四親本群體和 1個八親本群體。8個親本分別是SAGC-08 (A)、HHZ5-SAL9-Y3-Y1 (B)、BP1976B-2-3-7-TB-1-1 (C)、PR33282-B-8-1-1-1-1-1 (D)、FFZ1 (E)、CT 16658-5-2-2SR-2-3-6MP (F)、IR 68 (G)和IR 02A127 (H), 其來源及特點見表1。將8個親本分成ABCD和 EFGH 2組, 2組四親本間兩兩成對雜交(A×B、C×D 和 E×F、G×H), 產生 4對雙親本雜交種, 將4對雙親本雜交種F1再成對雜交產生2種四親本雜交種(A/B//C/D 和E/F//G/H), 2種四親本雜交種通過單粒傳法加代, 自交 6代, 建成 2個四親本MAGIC群體, 獲得221個穩定株系的A/B//C/D群體(記作DC1)和241個穩定株系的E/F//G/H群體(記作DC2)。分別從四親本雜交種 A/B//C/D和 E/F//G/H中選 25個 F1單株, 通過互交產生八親本雜交種(A/ B//C/D///E/F//G/H), 經過單粒傳法加代, 建成包含455個穩定株系的八親本MAGIC群體(記作DC3)。
1.2 田間設計與抗性鑒定
接種鑒定試驗在中國農業科學院作物科學研究所北京昌平實驗基地完成。2015年5月1日播種, 5月27日單本移栽, 株行距為20 cm ×17 cm。田間按2重復隨機區組設計, 每系每重復種植2行, 每行6株。全生育期田間采用常規管理, 僅治蟲, 不防病。
用于接種的白葉枯病菌為 2個遺傳穩定的菌系GD-V (強致病病菌)和C2 (弱致病菌)。首先將保存于-80℃的甘油中的菌種在 PSA培養基上復壯, 挑取單菌落經毒力測試后保存于 4℃。然后將篩選過的有毒菌株再培養于PSA培養基上于28℃培養48 h,進而配制成1×108~1×109cfu mL-1細菌懸浮液, 并于分蘗末期采用Kauffman剪葉法接種[14]。規定每個株系第1行的6株統一接種強致病菌GD-V, 第2行的6株接種弱致病菌C2; 從每株選取3~4張完全展開的葉接種。接種后保持田間有適度水, 以促進病斑擴展。待充分發病且病情穩定時(20 d)調查病斑長度。調查每重復6株, 測量每株 3張感病最長的葉片, 以這 3個表型值的平均值代表所調查單株的病斑長度, 每個株系的表型值為單個重復中所有單株病斑長度的均值。病斑長度<1 cm為高抗、1~5 cm為抗病、5~10 cm為中抗、10~15 cm為中感、>15 cm為感病。
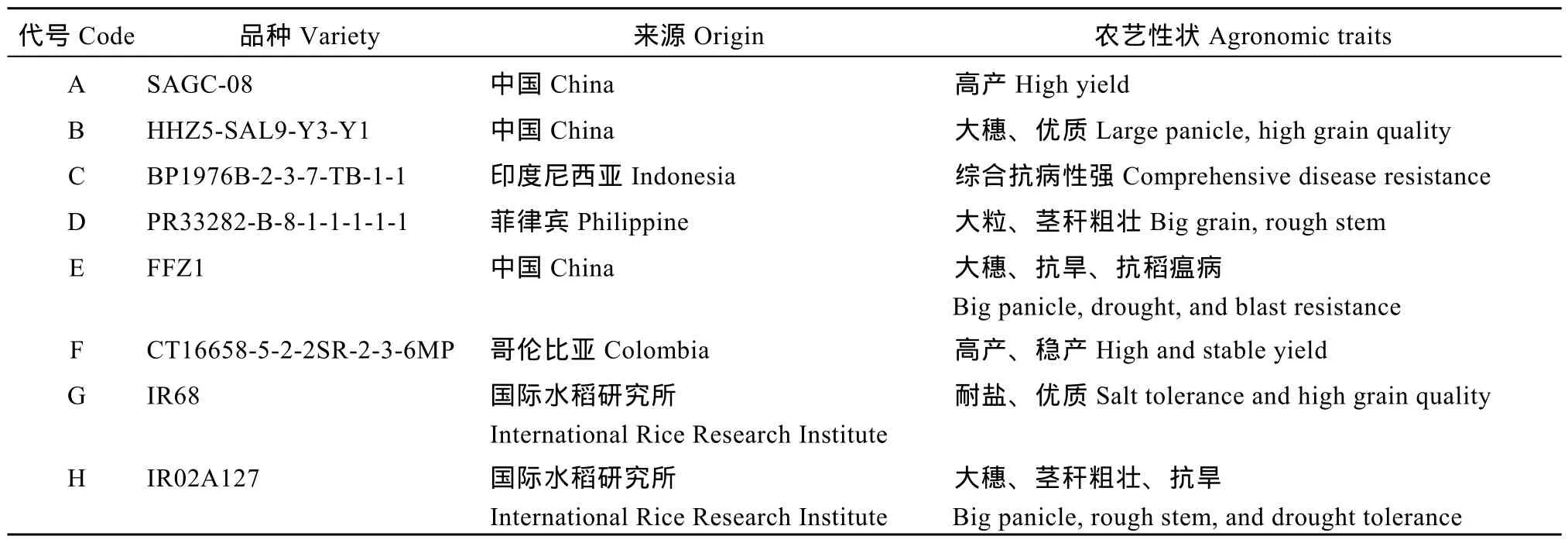
表1 用于構建MAGIC群體的品系來源及其主要特征Table1 Origin and agronomic characteristics of eight founder lines used in developing the MAGIC population
1.3 SNP基因型
DC1、DC2和 DC3群體的 DNA提取和基于Illumina Infinium技術的Rice 6K SNP芯片基因型分析在國際水稻研究所(International Rice Research Institute, IRRI)的基因型分析實驗室完成。Rice 6K SNP芯片由美國康奈爾大學Susan McCouch博士實驗室完成。按以下3個步驟過濾SNP基因型數據, 一是所有雜合基因型作為缺失數據, 刪除缺失 10%以上標記數據的株系; 二是剔除所有稀有等位基因頻率低于 3%的標記; 三是刪除標記間相關性很高(在0.95以上)的關聯標記。最終在 DC1、DC2和DC3群體中分別得到907、892和1329個高質量SNP位點用于群體結構分析和QTL定位研究。
1.4 群體結構分析
使用TASSEL V5.2.3軟件[15]評估群體結構和檢測位點間的連鎖不平衡(linkage disequilibrium, LD)水平。群體結構由主成分分析(principal components analysis, PCA)中的PC1和PC2來揭示。使用r2來衡量LD程度, 計算出所有可能位點組合的r2, 選用極顯著(P < 0.001)的位點對, 繪制LD衰減散點圖, 并繪制擬合曲線。以 LD衰減至起始值一半時所對應的物理距離作為LD衰減距離[13]。
1.5 數據分析和QTL定位
使用 SAS V9.2 (SAS Institute Inc., Cary NC, USA) PROCGLM分析不同基因型、不同菌系及菌系與基因型互作在不同群體中的方差組成, PROCCORR對不同菌系病斑長進行相關分析。利用 TASSEL V5.2.3軟件中的MLM (Mixed Linear Model)程序對SNP標記與表型值進行全基因組關聯分析, 以 P <0.001為閾值確定與白葉枯抗性顯著關聯的標記位點。
2 結果與分析
2.1 親本及MAGIC群體的抗病性表現
從 8個親本對 2個菌系的抗性差異比較(表2)可以看出, 無論是接種強致病菌還是弱致病菌, 8個親本間的抗性差異都達到極顯著水平。接種弱致病力菌系C2, 除親本B和E外, 其余親本都表現抗病。接種強致病力菌系GD-V, 8個親本對白葉枯病的敏病程度均顯著增加, 但親本D和G仍然表現中抗水平。總體而言, 來自中國的3個品種(A、B、E)對這2個致病菌系的抗性相對較差, 而來自東南亞和哥倫比亞的品種對2種菌株的抗性都相對較好。
對于3個MAGIC群體, 表型分析結果表明接種2個菌系各群體的病斑長度都表現出超親分離, 呈連續變異(表3和圖1)。因用于構建DC2群體及DC3群體的親本E中感C2菌系, 接種C2菌系后DC2和DC3群體的平均病斑長度顯著大于DC1群體; 接種菌系 GD-V, 不同群體的整體表現較為一致。從 3個群體病斑長度的頻率分布中可以看出接種C2菌系引起的病斑長度分布存在較高的峰, 表明各群體對 C2菌系的抗性受主基因控制, 但感病株系呈較大變異, 說明存在微效多基因的分離; 接種 GD-V型菌系, 3個群體病斑長度呈連續變異, 出現抗、感雙向均存在大幅度超親分離, 表現為典型的數量性狀遺傳。
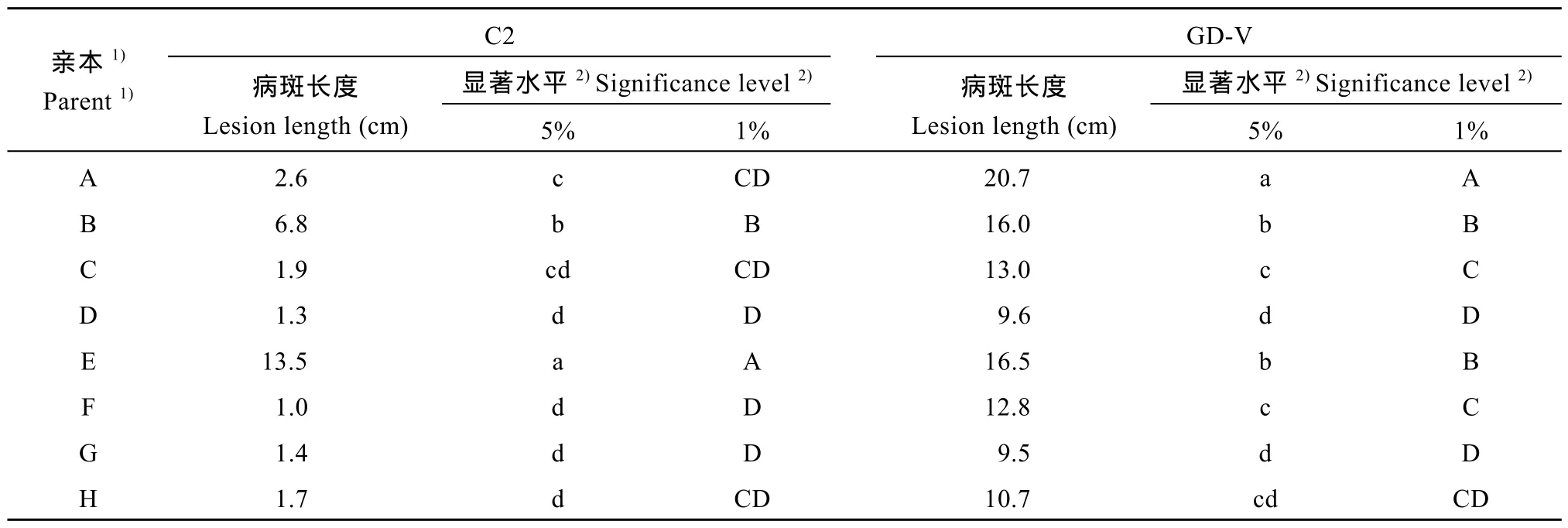
表2 親本對不同菌系的抗性差異比較Table2 Comparison of resistance to the Xoo strains among eight parents

表3 MAGIC群體接種白葉枯病菌后的病斑長度Table3 Lesion length (cm) of the MAGIC populations after inoculating two Xoo races
方差分析結果表明(表4), 重復間差異不顯著,不同菌系、不同基因型及菌系與基因型的互作均達到極顯著水平, 它們在DC1群體中分別解釋表型變異的66.04%、17.56%和11.95%, DC2群體中分別為45.78%、38.02%和 10.98%, DC3群體中分別為43.11%、39.24%和14.47%。相關分析表明, 接種強弱 2個致病菌后, 相同群體的病斑長度存在極顯著正相關, DC1群體接種C2和GD-V的相關系數為0.69, DC2群體為0.77, DC3群體為0.79, 表明多數株系對這2個菌系的抗性表現比較一致。
2.2 群體結構和連鎖不平衡分析
主成分分析結果表明, DC1群體的PC1和PC2分別是6.4%和5.0%, DC2群體的PC1和PC2分別是13.1%和3.0%, DC3群體的PC1和PC2分別是8.6% 和3.2%。所有群體沒有表現出明顯的群體結構。
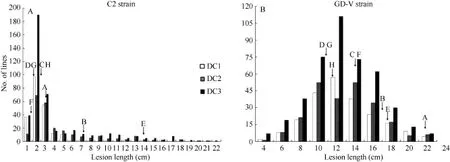
圖1 MAGIC群體接種C2 (A)和GD-V (B)菌株的病斑長度頻率分布Fig.1 Frequency distributions of lesion length caused by Xoo strain, C2 (A) and GD-V (B) in the MAGIC populations親本A~H的代號見表1 。Codes of A-H represent varieties given in Table1.
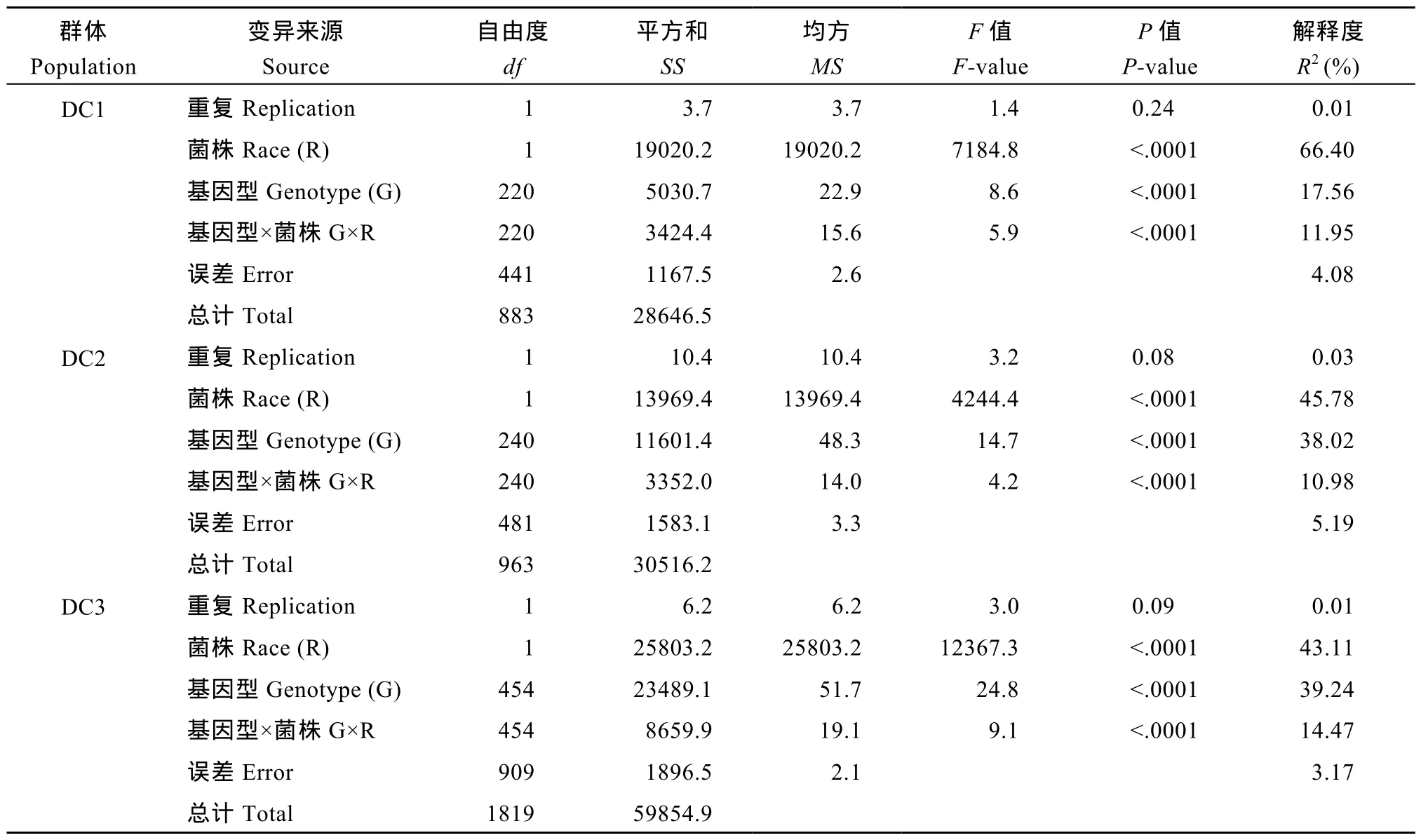
表4 MAGIC群體接種不同菌系病斑長度的方差組成Table4 Variance component estimates for multiple factors of lesion length caused by Xoo in the MAGIC populations
連鎖不平衡(LD)分析顯示3個群體的r2臨界值分別為0.14、0.20和0.08, 均表現出隨物理距離(Mb)增加而下降的趨勢, 且 DC3群體 LD衰減幅度比DC1和DC2群體快(圖2)。以LD降低至原始值的50% (r2> 0.2)作為LD的閾值, 3個群體LD衰減至一半的物理距離分別為 2.50、2.50和 1.25 Mb。從 r2臨界值和LD衰減幅度上來看, 8個親本群體DC3的連鎖不平衡程度最小, 衰減距離最短, 衰減速度最快, 而2個四親本群體DC1和DC2的連鎖不平衡表現趨于一致(圖2)。
2.3 白葉枯病抗性QTL定位
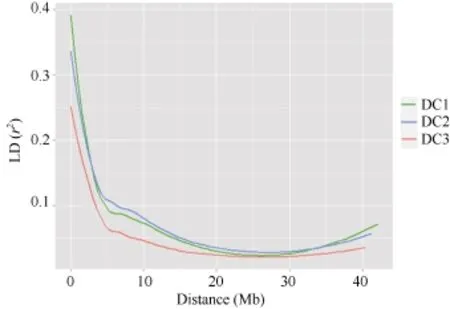
圖2 3個MAGIC群體的LD衰減Fig.2 LD (r2) decay of marker-pairs over all chromosomes as a function of physical distance (Mb) for three MAGIC populations
3 個群體中共檢測出7個抗白葉枯病QTL, 分布在第2、第6、第7、第9和第11染色體上(圖3和表5)。接種C2菌系, DC1群體共檢測出3個QTL, 即QBbr2-1、QBbr11-1和 QBbr11-2, 分別解釋表型變異的6.63%、9.45%和12.40%; DC2群體檢測出2個QTL,即QBbr11-1和QBbr11-2, 分別解釋表型變異的9.40% 和26.04%; DC3群體共檢測出3個QTL, 即QBbr6、QBbr11-1和QBbr11-2, 分別解釋表型變異的2.47%、9.75%和26.90%。接種GD-V菌系, DC1群體檢測出2 個QTL, 即QBbr2-2和QBbr9, 分別解釋表型變異的5.96%和6.32%; DC2群體檢測出2個QTL, 即QBbr11-1 和QBbr11-2, 分別解釋表型變異的4.82%和6.22%; DC3群體檢測出3個QTL, 即QBbr7、QBbr11-1和QBbr11-2,分別解釋表型變異的2.37%、2.61%和2.83%。
所有被檢測的 QTL中, 只有 QBbr11-1和QBbr11-2在不同群體中被檢測到對不同菌系都表現抗性, 但抗性效應存在顯著差異。顯然QBbr11-2對弱毒性 C2菌系表現出主基因抗性, 但對強毒性GD-V表現出數量抗性, 表明抗性QTL存在小種專化性, 這種專化性具體表現在同一抗病QTL對不同菌系的抗性反應不同及對不同菌系抗性效應存在差異。QBbr11-2在不同群體中對不同菌系均能被檢測到且效應都最大, 該位點的有利等位基因來自親本A、C、D、G和H。
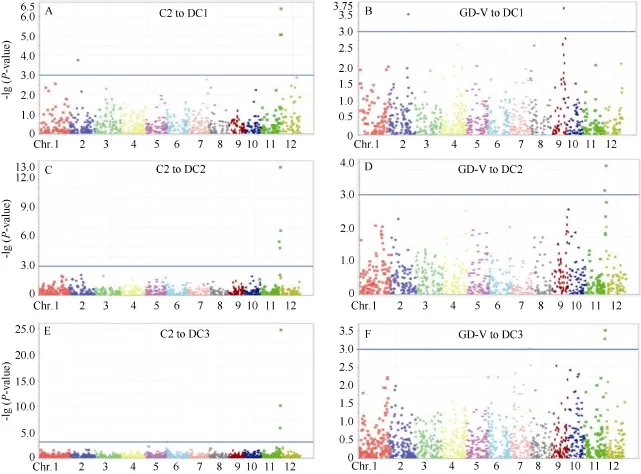
圖3 全基因組關聯分析定位影響MAGIC群體對2個白葉枯病菌的抗性QTLFig.3 Genome-wide association analysis of QTLs underlying resistance to two bacterial blight strains in MAGIC populations
2.4 抗病QTL定位的遺傳背景效應
2 個四親本群體DC1和DC2, 盡管它們的親本數目相同、群體大小和標記密度相近, 但親本的遺傳差異較大導致群體間的遺傳差異也較大。DC1群體共檢測出5個QTL, DC2群體共檢測出2個QTL,其中QBbr11-1和QBbr11-2在2個群體中都被檢測到。接種C2菌系, QBbr11-2在DC2群體中解釋表型變異的 26.04%, 為 DC1群體(12.40%)的 2倍多;接種GD-V菌系, QBbr11-1和QBbr11-2只在DC2群體中被檢測到, 表型貢獻率相似, 而在 DC1群體中均未被檢測到。相似地, 八親本群體相對四親本群體親本數目加倍, 群體大小也接近 2倍。比較四親本群體和八親本群體接種 C2菌系的定位結果發現,QBbr11-1和QBbr11-2在DC1、DC2和DC3群體中均被檢測到, QBbr11-1在3個群體中的表型貢獻率相似, 但QBbr11-2在DC1群體中的表型貢獻率幾乎是DC2和DC3群體中的一半。接種GD-V菌系時只在DC2和DC3群體中檢測到QBbr11-2, 對表型貢獻率均較小, 而在DC1群體中未檢測到該QTL。表明抗病QTL的表達具有明顯的遺傳背景效應, 具體體現在同一抗病QTL在不同群體中被檢測的效果不同, 而且表型效應也存在明顯差異。
2.5 抗病材料的篩選
根據 3個群體對強致病菌GD-V的抗性, 從 3個群體中共篩選出 8份抗病材料, 其中從 DC1和DC2群體各篩選出1份, DC3群體篩選出6份(表6)。這些材料接種弱致病力菌系C2的病斑長度除L1602 和L1959外均小于1 cm, 達到高抗水平; 接種強致病力菌系GD-V的病斑長度均小于4 cm, 達到抗病水平。通過對比這些株系攜帶的抗病位點可以看出,同一遺傳背景下如DC3群體, 不同株系的抗性水平與攜帶的抗病QTL數目之間具有較高的一致性, 而且帶有第11染色體QBbr11-2主基因的株系抗性較強, QBbr11-1與QBbr11-2互作表現出較強的抗性效應。表明對弱毒菌系通過主效抗病QTL與微效QTL的累加, 或對強毒菌系通過不同微效 QTL的累加,都可以顯著提高寄主對白葉枯病菌的抗性水平。
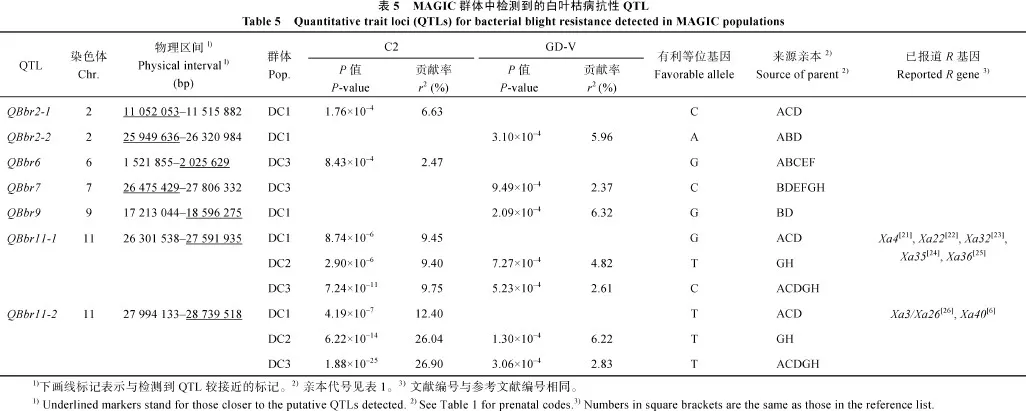
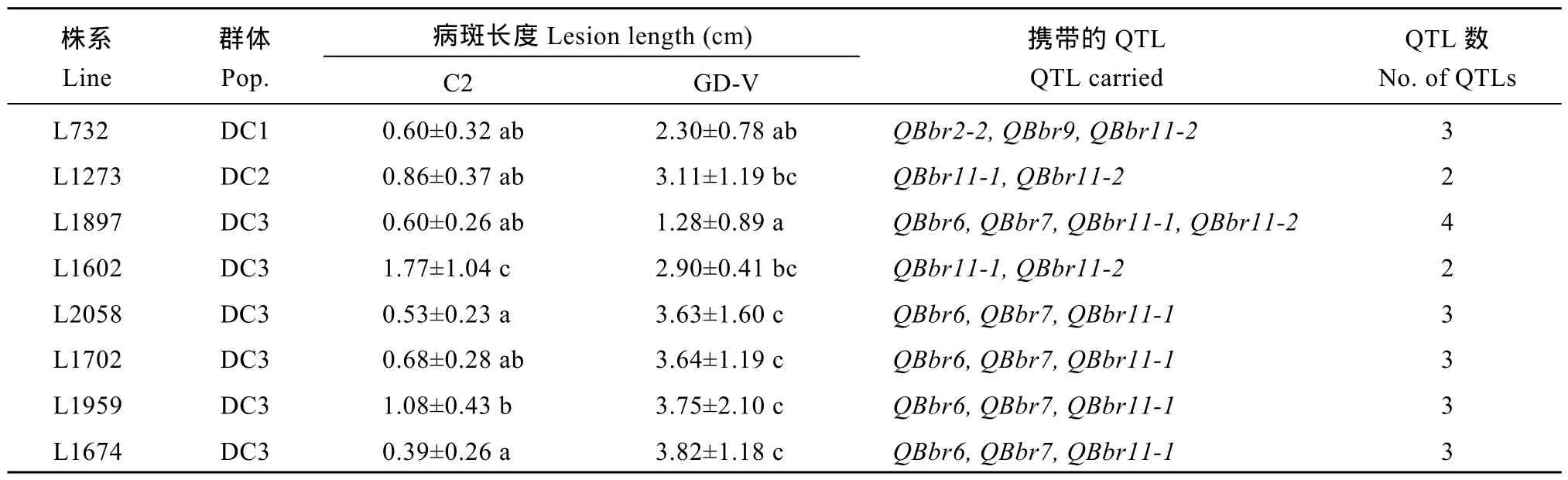
表6 MAGIC群體中篩選出抗病株系的抗性及抗病QTL的分布Table6 Lines exhibiting resistance to bacterial blight screened from MAGIC populations and distributions of resistant QTLs
3 討論
3.1 利用MAGIC群體定位QTL的優勢
與傳統的雙親本群體如RIL相比, MAGIC群體中更多的親本數量增加了等位基因多樣性, 大量累積的重組事件會提高被檢測QTL準確性, 并提高作圖分辨率[1 6-1 7]。與種質資源等自然群體相比,MAGIC群體降低了假陽性的出現并提高稀有等位基因檢測的效率。利用MAGIC群體定位QTL還具有三項優勢: (1)相比傳統雙親本定位群體, MAGIC群體中包含來源于不同親本的多個等位基因, 可以同時探討多個等位基因對某個性狀的影響。(2)以前通過雙親群體定位的很多QTL在離開了特定的遺傳背景后, 對相應的農藝性狀無效。MAGIC群體由不同親本來源的多個群體構成, 比較不同遺傳背景群體的定位結果, 就能發現受親本背景影響較小的QTL, 使QTL的效應能夠更加確定和具有通用性。如本研究中定位出的QBbr11-1和QBbr11-2在不同群體中均被檢測到, 說明這2個QTL受遺傳背景影響小, 可靠性較高, 可以用于進一步研究。(3)構建MAGIC群體可選用育種項目中性狀優異的材料作為親本, 多次重組創造大量的遺傳變異, 群體中出現的優良株系可用做育種中間材料或直接選育品種,定位到的QTL可以直接指導育種, 真正做到育種群體和定位群體的有機整合[18-20]。本研究篩選了 8份抗病株系, 聚合多個抗病 QTL, 不但可以用于進一步的遺傳研究, 還具有較高抗病育種價值。我們已經將部分抗病材料與感病材料配組, 構建主效抗病QTL的精細定位群體, 以期挖掘新的抗病基因或驗證本研究定位到的主效抗病QTL與已往報道的抗病基因或QTL的關系。
不可否認, 由于本研究所用芯片通量不高, 造成在每個群體中呈現多態的標記數量有限, 因而無法有效實現抗病位點的有利等位基因的剖析, 使得多親本群體在基因定位上的優越性受到一定程度的制約。有鑒于此, 我們正有利用3K重測序種質資源開發的56K芯片, 重新鑒定這3套多親本群體SNP基因型, 以實現基因或QTL的精細定位。
3.2 QTL定位結果的比較
根據與定位到的QTL顯著關聯SNP標記的物理位置, 我們將本研究定位到的抗水稻白葉枯病QTL與前人的定位結果比較, 發現第 11染色體QBbr11-1所在區域26 301 538~27 591 935存在已被定位的水稻白葉枯抗性基因 Xa4[21]、Xa22[22]、Xa32[23]、Xa35[24]和Xa36[25], 在QBbr11-2所在區域27 994 133~28 739 518存在Xa3/Xa26[26]和Xa40[6]基因。本研究結果表明QBbr11-1對弱毒力菌系C2和強毒力菌系 GD-V均表現為一個微效基因, 而QBbr11-2對C2表現為質量抗性基因, 但對GD-V仍有顯著的殘效, 表現為一個加性效應的微效 QTL。上述定位到的2個抗病QTL與所在區域已報道的白葉枯病抗性基因的關系, 還需加密標記精細定位后方能知曉。此外, 在第2、第6、第7和第9染色體上定位到的 5個抗病 QTL (QBbr2-1、QBbr2-2、QBbr6、QBbr7和 QBbr9)所在區域, 未發現有抗白葉枯病基因或QTL的報道, 可能屬于新發現的抗白葉枯病位點。
3.3 抗病QTL定位的遺傳背景效應
QTL表達的遺傳背景效應在水稻抗旱[27-28]、耐鹽[29-30]等性狀上已有報道。在植物抗病基因表達上也存在遺傳背景效應, Banerjee等[31]發現擬南芥抗細菌性斑點病菌(P.syringae)基因RPS2在Po-1遺傳背景下不起作用, 但該基因在哥倫比亞(Col-0)品種背景下對細菌性斑點病菌的 avrRpt2致病因子具有完全抗性。在水稻抗白葉枯病基因表達的遺傳背景方面, Xa21[32]和Xa26[33]的表達受遺傳背景影響, 認為這兩個抗性基因在粳稻背景下比在秈稻背景下抗性表達更完全。本研究中, 同樣2個四親本群體DC1 和DC2, 群體大小相似, 對C2和GD-V菌系DC1群體共檢測出5個QTL, 而DC2群體只檢測出2個QTL, 其中 QBbr2-1、QBbr2-2和 QBbr9只在 DC1中被檢測到, 未能在 DC2群體中檢測到。接種 C2菌系, 盡管QBbr11-1和QBbr11-2在2個群體中都被檢測到, QBbr11-2在 DC2群體中的表型變異(26.04%)比在DC1群體(12.40%)中高出2倍多。同樣, 接種 GD-V菌系, QBbr11-1和 QBbr11-2只在DC2群體中檢測到, 在DC1群體中未檢測到, 表明抗白葉枯病QTL存在明顯的遺傳背景效應。抗性基因表達存在遺傳背景效應, 意味著抗性基因從一個背景轉育到另一個完全不同的背景下, 其抗性水平可能會發生變化, 這種遺傳背景效應在抗病育種中應引起足夠的重視。
3.4 抗病QTL的育種應用
近幾十年來, 利用來自雙親構建的重組自交系、DH系、雙向導入系等群體定位了大量水稻抗白葉枯病QTL[34-37], 但多數QTL效應不大, 有些與環境間有很強的互作, QTL表達的遺傳背景效應明顯,育種應用價值較小。一直以來, 對水稻白葉枯病的遺傳研究和抗性育種主要集中在主基因抗性[37]。迄今雖然定位到了30多個質量抗性基因, 但抗病育種大多集中利用Xa4、Xa21、Xa23等少數廣譜抗性基因[7,38]。這種抗性基因的集中利用會導致白葉枯病菌的消長, 產生新的致病菌系, 造成對帶有 Xa4基因的品種在中國和帶有Xa21基因的品種在菲律賓、韓國、印度和中國喪失抗性[39-41]。因此, 發掘新的抗性基因或將不同抗性基因累加是提高品種的抗性水平或延長抗病品種使用年限的重要舉措。本研究表明, 水稻對白葉枯病存在主基因抗性和多個微效基因抗性, 它們共同影響抗性水平, 這與3個多親本群體中觀察到抗性的極端超親分離相符合。鑒于水平抗性數量化及抗病 QTL小種專化性弱的特點, 利用標記輔助選擇方法借助緊密連鎖的分子標記進行質量抗性基因和水平抗性QTL的組合, 可能是提高品種抗性水平的有效措施[42]。本研究定位了來自不同親本的 7個抗性 QTL, 大多表現為數量抗性, 具有明顯的抗性聚合效應。將篩選出的 8個不同抗病QTL的聚合株系作抗性供體, 通過分子標記輔助選擇將這些抗性基因導入到優良品種背景, 可以培育出符合育種目標的抗病品種。
4 結論
在遺傳上相互關聯的2個四親本群體和1個八親本群體中觀察到對白葉枯病弱毒菌系 C2和強毒菌系GD-V的抗性超親分離。共檢測到影響水稻白葉枯病抗性的7個QTL, 大多QTL均表現數量抗性,抗性表達存在明顯的遺傳背景效應。QBbr11-2對C2表現質量抗性, 對GD-V表現一定的抗性殘余效應。QBbr11-1和QBbr11-2受遺傳背景影響較小, 具有一定的育種應用價值。從3個群體篩選出8份不同抗病QTL聚合的抗病材料, 可用作抗病育種的中間抗源。本研究證實了水稻多親本群體既是遺傳研究群體也是理想的育種群體, 可以實現基因定位與育種應用的有機結合。
致謝: 感謝中國農業科學院作物科學研究所水稻分子遺傳與分子育種實驗室周永力老師和王明明同學幫助培養菌株, 張強、張建、王小倩等同學幫助田間接種和病斑調查。
References
[1] Mew T W.Current status and future prospects of research on bacterial blight of rice.Annu Rev Phytopathol, 1987, 25: 359-382
[2] Chen S, Liu X, Zeng L, Ou-Yang D M, Yang J, Zhu X.Genetic analysis and molecular mapping of a novel recessive gene xa34(t)for resistance against Xanthomonas oryzae pv.oryzae.Theor Appl Genet, 2011, 122: 1331-1338
[3] Ni?o-Liu D O, Ronald P C, Bogdanove A J.Xanthomonas oryzae pathovars: model pathogens of a model crop.Mol Plant Pathol,2006, 7: 303-324
[4] 虞玲錦, 張國良, 丁秀文, 高雨, 謝寅峰.水稻抗白葉枯病基因及其應用研究進展.植物生理學報, 2012, 48: 223-231 Yu L J, Zhang G L, Ding X W, Gao Y, Xie Y F.Progress in identification and application of resistance genes to bacterial blight.Plant Physiol J, 2012, 48: 223-231 (in Chinese with English abstract)
[5] 羅利軍, 梅捍衛, 趙新華, 鐘代彬, 王一平, 余新橋, 應存山.水稻白葉枯病抗性基因定位及其小種專化性.中國科學, 1998,28: 536-541 Luo L J, Mei H W, Zhao X H, Zhong D B, Wang Y P, Yu X Q,Ying C S.Mapping of resistance genes to bacterial blight of riceand their race specificity in rice.Chin Sci, 1998, 28: 536-541 (in Chinese)
[6] Kim S M, Suh J P, Qin Y, Noh T H, Reinke R F, Jena K K.Identification and fine-mapping of a new resistance gene, Xa40, conferring resistance to bacterial blight races in rice (Oryza sativa L.).Theor Appl Genet, 2015, 128: 1933-1943
[7] 章琦.中國雜交水稻白葉枯病抗性的遺傳改良.中國水稻科學, 2009, 23: 111-119 Zhang Q.Genetics and improvement of resistance to bacterial blight in hybrid rice in China.Chin J Rice Sci, 2009, 23: 111-119 (in Chinese with English abstract)
[8] Khan M A, Naeem M, Iqbal M.Breeding approaches for bacterial leaf blight resistance in rice (Oryza sativa L.), current status and future directions.Eur J Plant Pathol, 2014, 139: 27-37
[9] Person C, Samborski D J, Rohringer R.The gene-for-gene concept.Nature, 1962, 194: 561-562
[10] Ea V D B, Jones J D.Plant disease-resistance proteins and the gene-for-gene concept.Trends Biolchem Sci, 1998, 23: 454-456
[11] Li Z K, Luo L J, Mei H W, Paterson A H, Zhao X H, Zhong D B,Wang Y P, Yu X Q, Zhu L, Tabien R, Stansel J W, Ying C S.A “defeated” rice resistance gene acts as a QTL against a virulent strain of Xanthomonas oryzae pv.oryzae.Mol Gen Genet, 1999,261: 58-63
[12] Zhao K Y, Tung C W, Eizenga G C, Wright M H, Ali M L, Price A H, Norton G J, Islam M R, Reynolds A, Mezey J, McClung A M, Bustamante C D, McCouch S R.Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa.Nat Commun, 2011, 2: 1-10
[13] Meng L J, Zhao X Q, Ponce K, Leung H, Ye G Y.QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines.Field Crops Res, 2016, 189: 19-42
[14] Kauffman H E, Reddy A P K, Hsieh S P Y, Merca S D.Improved technique for evaluating resistance of rice varieties to Xanthomonas oryzae.Plant Dis Rep, 1973, 57: 537-541
[15] Bradbury P, Zhang Z, Kroon D, Casstevens T Y, Buckler E.TASSEL: software for association mapping of complex traits in diverse samples.Bioinformatics, 2007, 23: 2633-2635
[16] Valdar W, Flint J, Mott R.Simulating the collaborative cross:Power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice.Genetics,2006, 172: 1783-1797
[17] Cavanagh C, Morell M, Mackay I, Powell W.From mutations to MAGIC, resources for gene discovery, validation and delivery in crop plants.Curr Opin Plant Biol, 2008, 11: 215-221
[18] Huang B E, Verbyla K L, Verbyla A P, Raghavan C, Singh V K,Gaur P, Leung H, Varshney R K, Cavanagh C R.MAGIC populations in crops: current status and future prospects.Theor Appl Genet, 2015, 128: 999-1017
[19] Bandillo N, Raghavan C, Muyco P A, Sevilla M A L, Lobina I T,Dilla-Ermita C J, Tung C W, McCouch S, Thomson M, Mauleon R, Singh R K, Gregorio G, Redo?a E, Leung H.Multi-parent advanced generation inter-cross (MAGIC) populations in rice:progress and potential for genetics research and breeding.Rice,2013, 6: 1-15
[20] Leung H, Raghavan C, Zhou B, Oliva R, Choi I R, Lacorte V, Jubay M L, Cruz C V, Gregorio G, Singh R K, Ulat V J, Borja F N, Mauleon R, Alexandrov N N, McNally K L, Hamilton R S.Allele mining and enhanced genetic recombination for rice breeding.Rice, 2015, 8: 1-11
[21] Sun X, Yang Z, Wang S, Zhang Q.Identification of a 47-kb DNA fragment containing Xa4, a locus for bacterial blight resistance in rice.Theor Appl Genet, 2003, 106: 683-687
[22] Wang C T, Tan M P, Xu X, Wen G S, Zhang D P, Lin X H.Localizing the bacterial blight resistance gene, Xa22(t), to a 100-kilobase bacterial artificial chromosome.Phytopathology,2003, 93: 1258-1262
[23] 鄭崇珂, 王春連, 于元杰, 梁云濤, 趙開軍.水稻抗白葉枯病新基因 Xa32(t)的鑒定和初步定位.作物學報, 2009, 35:1173-1180 Zheng C K, Wang C L, Yu Y J, Liang Y T, Zhao K J.Identification and molecular mapping of Xa32(t), a novel resistance gene for bacterial blight (Xanthomonas oryzae pv.oryzae) in rice.Acta Agron Sin, 2009, 35: 1173-1180 (in Chinese with English abstract)
[24] 郭嗣斌, 張端品, 林興華.小粒野生稻抗白葉枯病新基因的鑒定與初步定位.中國農業科學, 2010, 43: 2611-2618 Guo S B, Zhang D P, Lin X H.Identification and mapping of a novel bacterial blight resistance gene Xa35(t) originated from oryza minuta.Sci Agric Sin, 2010, 43: 2611-2618 (in Chinese with English abstract)
[25] 苗麗麗, 王春連, 鄭崇珂, 車晉英, 高英, 溫義昌, 李貴全, 趙開軍.水稻抗白葉枯病新基因的初步定位.中國農業科學,2010, 43: 3051-3058 Miao L L, Wang C L, Zheng C K, Che J Y, Gao Y, Wen Y C, Li G Q, Zhao K J.Molecular mapping of a new gene for resistance to rice bacterial blight.Sci Agric Sin, 2010, 43: 3051-3058 (in Chinese with English abstract)
[26] Xiang Y, Cao Y L, Xu C G, Li X H, Wang S P.Xa3, conferring resistance for rice bacterial blight and encoding a receptor kinase-like protein, is the same as Xa26.Theor Appl Genet, 2006,113: 1347-1355
[27] Wang Y, Zhang Q, Zheng T Q, Cui Y R, Zhang W Z, Xu J L, Li Z K.Drought-tolerance QTLs commonly detected in two sets of reciprocal introgression lines in rice.Crop & Pasture Sci, 2014, 65:171-184
[28] Wang Y, Zang J P, Sun Y, Ali J, Xu J L, Li Z K.Background-independent quantitative trait loci for drought tolerance identified using advanced backcross introgression lines in rice.Crop Sci, 2013, 53: 430-441
[29] Cheng L R, Wang Y, Meng L J, Hu X, Cui Y R, Sun Y, Zhu L H,Ali J, Xu J L, Li Z K.Identification of salt-tolerant QTLs with strong genetic background effect using two sets of reciprocal introgression lines in rice.Genome, 2012, 55: 45-55
[30] 楊靜, 孫勇, 程立銳, 周政, 王韻, 朱苓華, 蒼晶, 徐建龍, 黎志康.利用雙向導入系群體檢測遺傳背景對耐鹽QTL定位的影響.作物學報, 2009, 35: 974-982 Yang J, Sun Y, Cheng L R, Zhou Z, Wang Y, Zhu L H, Cang J, Xu J L, Li Z K.Genetic background effect on QTL mapping for salt tolerance revealed by a set of reciprocal introgression line populations in rice.Acta Agron Sin, 2009, 35: 974-982 (in Chinese with English abstract)
[31] Banerjee D, Zhang X, Bent A F.The leucine-rich repeat domaincan determine effective interaction between RPS2 and other host factors in Arabidopsis RPS2-mediated disease resistance.Genetics, 2001, 158: 439-450
[32] 徐建龍, 林貽滋, 張炳林, 翁錦屏.水稻白葉枯病抗性基因Xa-21的初步利用.浙江農業學報, 1996, 8: 70-73 Xu J L, Lin Y Z, Zhang B L, Weng J P.Study on utilization of Xa-21 gene resistant to bacterial blight in rice.Acta Agric Zhejianggensis, 1996, 8: 70-73 (in Chinese with English abstract)
[33] Sun X L, Gao Y L, Yang Z F, Xu C G, Li X H, Wang S P, Zhang Q F.Xa26, a gene conferring resistance to Xanthomonas oryzae pv.oryzae in rice, encodes an LRR receptor kinase-like protein.Plant J, 2004, 37: 517-527
[34] 于彥春, 滕勝, 曾大力, 董國軍, 錢前, 黃大年, 朱立煌.水稻抗白葉枯病微效 QTL的定位分析.中國水稻科學, 2003, 17:315-318 Yu Y C, Teng S, Zeng D L, Dong G J, Qian Q, Huang D N, Zhu L H.Analysis of QTLs for resistance to rice bacterial bight.Chin J Rice Sci, 2003, 17: 315-318 (in Chinese with English abstract)
[35] Wang C M, Su C C, Zhai H Q, Wan J M.Identification of QTLs underlying resistance to a virulent strain of Xanthomonas oryzae pv.oryzae in rice cultivar DV85.Field Crops Res, 2005, 91:337-343
[36] Li Z K, Arif M, Zhong D B, Fu B Y, Domingo-Rey J, Ali J,Vijayakumar C H M, Yu S B, Khush G S.Complex genetic networks underlying the defensive system of rice (Oryza sativa L.)to Xanthomonas oryzae pv.oryzae.Proc Natl Acad Sci USA, 2006, 103: 7994-7999
[37] Zhang F, Xie X W, Xu M R, Wang W S, Xu J L, Zhou Y L, Li Z K.Detecting major QTL associated with resistance to bacterial blight using a set of rice reciprocal introgression lines with high density SNP markers.Plant Breed, 2015, 134: 286-292
[38] Zhou Y L, Uzokwe V N E, Zhang C H, Cheng L R, Wang L,Chen K, Gao X Q, Sun Y, Chen J J, Zhu L H, Zhang Q, Ali J, Xu J L, Li Z K.Improvement of bacterial blight resistance of hybrid rice in China using the Xa23 gene derived from wild rice (Oryza rufipogon).Crop Protection, 2011, 30: 637-644
[39] Lee S W, Choi S H, Han S S, Lee D G, Lee B Y.Distribution of Xanthomonas oryzae pv.oryzae strains virulent to Xa21 in Korea.Phytopathology, 1999, 89: 928-933
[40] Shanti M L, George M L C, Cruz C M V, Bernardo M A, Nelson R J, Leung H,Reddy J N, Sridhar R.Identification of resistance genes effective against rice bacterial blight pathogen in eastern India.Plant Dis, 2001, 85: 506-512
[41] 曾列先, 黃少華, 伍尚忠.IRBB21 (Xa21)對廣東稻白葉枯病菌5個小種的抗性反應.植物保護學報, 2002, 29: 97-100 Zeng L X, Huang S H, Wu S Z.Resistance of IRBB21 (Xa21) to five races of bacterial blight in Guangdong.Acta Phytophyl Sin,2002, 29: 97-100 (in Chinese)
[42] Gu K Y, Yang B, Tian D S, Wu L F, Wang D J, Sreekala C,Yang F, Chu Z Q, Wang G L, White F F, Yin Z C.R gene expression induced by a type-III effector triggers disease resistance in rice.Nature, 2005, 435: 1122-1125
Mapping of QTLs for Bacterial Blight Resistance and Screening of Resistant Materials Using MAGIC Populations of Rice
CHEN Tian-Xiao1,2, ZHU Ya-Jun3, MI Xue-Fei3, CHEN Kai3, MENG Li-Jun3, ZUO Shi-Min1,*, and XU Jian-Long2,3,4,*
1Key Laboratory of Plant Functional Genomics of Jiangsu Province / Key Laboratory of Crop Genetics and Physiology of Jiangsu Province, Yangzhou University, Yangzhou 225009, China;2Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081, China;3Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518210, China;4Shenzhen Institute of Breeding & Innovation, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China
Three genetically interconnected multi-parents advanced generation inter-cross (MAGIC) population, including two populations (DC1 and DC2) derived from four parents and one population from eight parents (DC3) were used to detect QTLs for resistance to two strains, a weak virulent C2 and a strong virulent GD-V of Xanthomonas oryzae pv.oryzae (Xoo) and to screen resistant breeding materials.Most parents were resistant to C2 and susceptible to GD-V.Transgressive segregations of lesion length for the two strains were observed in the three MAGIC populations and showed continuous distributions.A total of seven QTLs affecting lesion length of two strains were detected.Most QTLs showed quantitative resistance and obvious genetic background effect.Among the seven QTLs, QBbr11-1 and QBbr11-2 had less genetic background effect, which is valuable in ricebreeding for disease resistance.Eight resistant lines pyramiding different QTLs were screened from the three MAGIC populations,indicating the combination of qualitative resistance gene and quantitative resistance gene can significantly improve resistance level.The eight resistant breeding lines could be used as resistant donors in rice breeding for resistance.The results indicated that the MAGIC populations are ideal material for genetic study and marker-assisted breeding, showing a tight integration of genetic research and breeding application in rice.
Multi-parent Advanced Generation Inter-Crosses (MAGIC); Rice bacterial blight; Quantitative trait loci (QTL); Genome-wide association study (GWAS); Rice
10.3724/SP.J.1006.2016.01437
本研究由國家高技術研究發展計劃(863計劃)項目(2014AA10A601), 深圳孔雀團隊計劃項目(20130415095710361), 中國農業科學院科技創新工程團隊和江蘇重點研發計劃(現代農業)(BE2015363)項目資助。
This study was supported by National High-Tech Research & Development Plan (863 Program) (2014AA10A601), Shenzhen Peacock Plan (20130415095710361), Scientific and Technological Innovation Project of Chinese Academy of Agricultural Sciences, and the Key Research Plan of Modern Agriculture of Jiangsu Province (BE2015363).
(Corresponding authors): 徐建龍, E-mail: xujlcaas@126.com, Tel: 010-82105854; 左示敏, E-mail: smzuo@yzu.edu.cn, Tel:0514-87972136
聯系方式: E-mail: chentx2006@163.com, Tel: 010-82105855
Received(): 2016-03-07; Accepted(接受日期): 2016-06-20; Published online(網絡出版日期): 2016-07-04.
URL: http://www.cnki.net/kcms/detail/11.1809.S.20160704.0826.012.html
猜你喜歡
幼兒100(2023年39期)2023-10-23 11:36:32
青少年科技博覽(中學版)(2022年6期)2022-12-27 19:44:27
中國土壤與肥料(2021年5期)2021-12-12 02:02:11
今日農業(2021年21期)2021-11-26 05:07:00
軍事文摘(2021年22期)2021-11-26 00:43:51
今日農業(2021年14期)2021-10-14 08:35:40
金橋(2021年7期)2021-07-22 01:55:38
今日農業(2020年20期)2020-11-26 06:09:10
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
