在線測定秦皮甲素和秦皮乙素水解反應速率常數的ど集擦鞫注射芯片毛細管膠束電動色譜方法
2016-11-19 19:17:21朱華東陳永雷陳宏麗陳興國
分析化學 2016年4期
朱華東 陳永雷 陳宏麗 陳興國

摘 要 建立了在線測定秦皮甲素和秦皮乙素水解反應速率常數的掃集流動注射膠束電動色譜新方法。該方法進樣頻率為12次/h,可在5 min內完全分離反應體系中的所有組分,30 min即可完成一個溫度下的反應速率常數測定。由于該在線測定方法不需要終止水解反應,通過一次連續進樣分析得到水解反應過程電泳譜圖,從而可以獲得水解反應過程中的一些信息。在最佳條件時,用0.1 mol/L KOH為催化劑,測得25, 30, 35, 40和45℃時秦皮甲素的水解反應速率常數分別為3.65×10
1 引 言
秦皮作為一種常用的中藥,已經有2000多年的歷史,對治療腹瀉、痢疾、咳嗽和一些婦科疾病有很好的療效,并且有一定的防癌作用[1,2]。秦皮甲素(Aesculin)和秦皮乙素(Aesculetin)是秦皮的兩種主要有效成分,其結構如圖1所示。藥理實驗表明,秦皮甲素可以延長環己烯巴比妥對小鼠的催眠作用,而秦皮乙素則沒有此作用;秦皮甲素也不具有秦皮乙素的抗組胺作用以及放松對豚鼠離體氣管平滑肌的作用。由于它們在潮濕的環境中性質不穩定,因此建立一種簡單、經濟、可靠的研究秦皮甲素和秦皮乙素的水解方法對此類藥物的質量和穩定性評價具有重要意義。
毛細管電泳(Capillary electrophoresis, CE)由于具有所需樣品量小、分析時間短和分離效率高的優點而適用于化學反應動力學參數的測定[3~10]。Zhang等[7]利用離線CE方法測定了秦皮甲素堿性水解反應速率常數和活化能。該方法雖然可行,但存在檢測反應液中目標化合物需先用磷酸溶液終止反應、每次實驗只能得到所需要測定的動力學參數中的一個點等缺點。在測定速率常數時,至少需要對反應過程中5個不同反應時間點的反應液進行分析。若欲測定不同溫度、pH值、離子強度、溶劑等條件下的動力學參數,可能需要幾十甚至上百次實
圖1 秦皮甲素和秦皮乙素的化學結構式
Fig.1 Chemical structure of aesculin and aesculetin
驗,這給反應動力學研究帶來了極大不便。離線分析表明秦皮甲素水解生成秦皮乙素和葡萄糖。從這一結果看,秦皮甲素的內酯鍵在堿性條件下似乎是穩定的。然而,Chen等[11]的研究表明,烯醇式和內酯結構很容易在堿性條件下解離。此外,Dodge[12]指出,香豆素的堿性水解和其內酯形成是一個可逆過程,當堿過量時,水解完全。但是,過量的堿一旦被中和,則開始進行逆水解過程,即開始形成香豆素,直到達到平衡點。很明顯離線分析只給出了水解反應終止后的秦皮甲素水解反應在平衡狀態的信息,而無法獲得亞穩狀態的一些信息。而這些信息對研究秦皮甲素類藥物的物理、化學、生物性質的各個過程可能是非常重要的。迄今為止,測定秦皮乙素水解反應速率常數的工作尚未見報道。由此可知,有必要建立在線研究秦皮甲素和秦皮乙素水解反應的新方法。
自從1997年Kubán[ 13]和Fang[14]等分別建立了流動注射毛細管電泳(FIchip CE)技術以來,FIchip CE已經廣泛應用于各種分析領域[15~24], 并在此基礎上形成了流動注射芯片毛細管電泳(FIchip CE)。由于具有高樣品吞吐量和連續進樣等優點,FIchip CE非常適用于化學反應動力學研究。然而,迄今為止,該技術很少用于水解動力學研究中。
鑒于此,本研究建立了一種簡單快速的在線測定水解反應速率常數的掃集流動注射毛細管膠束電動色譜(SweepingFIchip MEKC)方法, 并將其用于測定秦皮甲素和秦皮乙素的水解反應速率常數。
2 實驗部分
2.1 試劑
秦皮甲素和秦皮乙素均購于中國藥品生物制品檢定所。十六烷基三甲基溴化銨(CTAB)、十二烷基硫酸鈉(SDS)、Na2B4O7、KH2PO4、HCl、KOH和乙腈(ACN)購于天津第二試劑廠。所有試劑均為分析純。所用水均為蒸餾水。
2.2 FIchip CE系統
用于在線研究秦皮甲素和秦皮乙素水解反應的裝置由K1000 FIA型流動注射分析儀(HITACHI,日本)、H通道微芯片聯用接口和HPE100型電泳儀(BioRad 公司,美國)組成,整個裝置如圖2所示。用一根33 cm的PTFE (0.5 mm I.D.)泵管連接FI的進樣閥和分流接口。通過FI的蠕動泵流路將緩沖溶液或水輸送到位于試劑環兩邊的兩個20 μL的試劑環中,將樣品溶液輸送位于中間的樣品環中。采用手動進樣模式。用恒溫水浴控制水解反應溫度,用磁力攪拌器混合反應物,置于恒溫水浴中的10 mL燒瓶作為反應器與輸送載流溶液(即運行緩沖溶液)的柱塞泵相連接。用有效長度為22.0 cm (總長25.0 cm)、內徑75 μm的熔融石英毛細管作為分離通道(河北永年光導纖維廠)。檢測波長為334 nm。用Chroma 軟件(BioRad 公司,美國)采集處理電泳數據。
.
2.3 樣品和試劑制備
用10%甲醇配制秦皮甲素(1.00 mmol/L)和秦皮乙素(0.80 mmol/L)儲備溶液,保存于4℃冰箱內備用。用10%甲醇稀釋上述儲備液, 得到濃度分別為0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.70, 0.80, 0.90和1.00 mmol/L秦皮甲素標準溶液和0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.70和0.80 mmol/L秦皮乙素標準溶液。分別配制0.1 mol/L Na2B4O7、0.2 mol/L KH2PO4、0.04 mol/L CTAB和0.2 mol/L SDS儲備溶液。分離緩沖溶液為5 mmol/L Na2B4O710 mmol/L KH2PO42.0 mmol/L CTAB5% ACN (pH 6.0)和10 mmol/L Na2B4O720 mmol/L KH2PO45.0 mmol/L SDS (pH 6.0)。所有溶液均用0.45 μm濾膜(上海新亞凈化器件廠)過濾。
2.4 實驗操作步驟
2.4.1 FI和CE操作步驟 采用16通自動進樣閥完成FI充樣(Loading)和注樣(Injecting),具體操作參見文獻[17]。
為保證良好的重現性,每天運行前分別用水、0.1 mol/L NaOH、水、緩沖溶液依次沖洗毛細管5 min。兩次運行之間毛細管依次用水(2 min)、0.1 mol/L NaOH (3 min)、水(2 min)、緩沖溶液(3 min)沖洗。
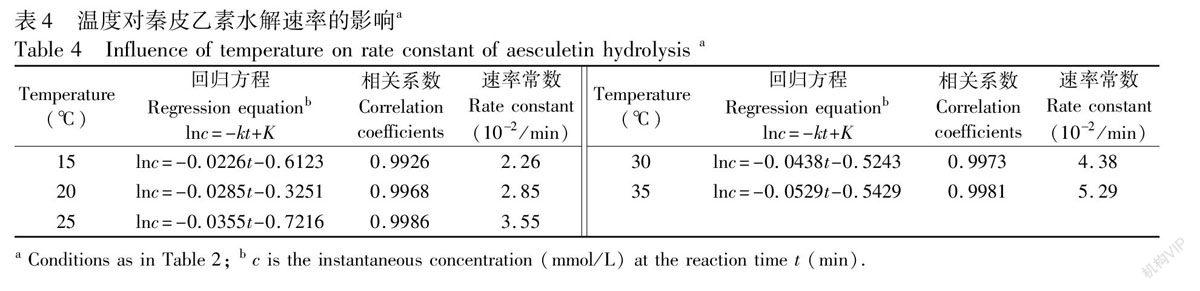
2.4.2 水解反應速率常數測定步驟 先將恒溫水浴調節到水解所需的溫度(分別為25, 30, 35, 40和45℃)。再將1.0 mL 1.00 mmol/L秦皮甲素溶液和1.0 mL 0.2 mol/L KOH溶液分別在恒溫水浴中預熱至相應溫度并恒溫15 min。然后將這兩種溶液用磁力攪拌器快速混合,同時,取反應混合溶液進樣并計時,進行分離、測定。此后每隔5 min進樣一次并進行分離測定。然后按同樣步驟分別在15, 20, 25, 30和35℃時測定秦皮乙素的水解反應速率常數。
3 結果與討論
3.1 CE條件優化
為了依據FIchip CE體系測得的數據研究秦皮甲素和秦皮乙素的水解速率常數,就必須要求CE能將二者的水解產物與共存物質完全分離。為此,必須對CE條件進行優化。
本實驗先用5 mmol/L Na2B4O710 mmol/L KH2PO4 (pH 6.0)作緩沖溶液[7]。結果表明,在此條件下只能得到兩個“矮胖”峰(電泳圖未給出)。其原因是樣品溶液中含有0.1 mol/L KOH,其電導率比緩沖溶液高得多,發生了去堆積效應。該效應可以通過在緩沖溶液中加入CTAB以形成膠束電動色譜來消除[25]。實驗考察了CTAB濃度在1.0~5.0 mmol/L范圍內變化時對峰高和峰形的影響。結果表明,當其濃度為2.0 mmol/L時,峰形和峰高最好。為了進一步改善分離效率,實驗還考察了乙腈作添加劑的可行性和其濃度的影響。結果表明,用乙腈作添加劑可有效提高分離效率,且當乙腈濃度為5%時,峰形最好。
FIchip CE的最大采樣頻率取決于水解反應時間和CE的分離速度,短的分離時間和高樣品吞吐量將有利于快速動力學研究。為此,實驗考察了電壓對分離時間和采樣頻率的影響。結果表明,雖然使用高電壓會提高分離速度、縮短分離時間,但同時會增加基線噪聲,使檢出限變高。當電壓為
Symbolm@@ 6.0 kV時(CTAB使EOF逆轉,需要采用負高壓),秦皮甲素和其水解產物在5 min內可得到完全分離,但是當電壓大于
Symbolm@@ 6.5 kV時,秦皮甲素和其水解產物的峰發生部分重疊。所以用
Symbolm@@ 6.0 kV作為最佳電壓。
在研究秦皮乙素的水解反應時,仍先用5 mmol/L Na2B4O710 mmol/L KH2PO4 (pH 6.0)作緩沖溶液,結果表明在此條件無法將目標分析物分離。隨后采用10 mmol/L Na2B4O720 mmol/L KH2PO4 (pH 6.0)為緩沖溶液,此時,采用文獻[7]中使用的0.4 mmol/L的初始反應物濃度時,秦皮乙素的峰高為2.1 mAU,水解10 min后幾乎檢測不到秦皮乙素的峰。這是由于使用紫外檢測器的FIchip CE低靈敏度導致的。為解決這一問題,實驗采用在線“掃集”技術[25~29],即建立了在線掃集流動注射芯片膠束電動色譜(SweepingFIchip MEKC)體系。具體步驟如下,用100 μL的樣品環和1.0 mL/min的流速增加進樣體積,在緩沖溶液中加入SDS形成膠束并實現MEKC分離。由于樣品溶液中不含SDS,施加電壓后SDS膠束將樣品區帶掃集到一個狹窄的區帶,從而達到富集效果。實驗考查了SDS濃度在5~30 mmol/L范圍內變化時對靈敏度的影響,結果表明,SDS的濃度為10 mmol/L時靈敏度最高。在此條件下,0.4 mmol/L秦皮乙素的峰高為22.5 mAU,靈敏度提高了10.7倍。因此,選擇10 mmol/L Na2B4O720 mmol/L KH2PO410 mmol/L SDS (pH 6.0)作為運行緩沖溶液。當電壓為6.5 kV時,秦皮乙素和其水解產物在5 min內可很好地分離。當電壓大于6.5 kV時,基線噪聲變差,分離度明顯下降。因此選擇6.5 kV作為最佳分離電壓。
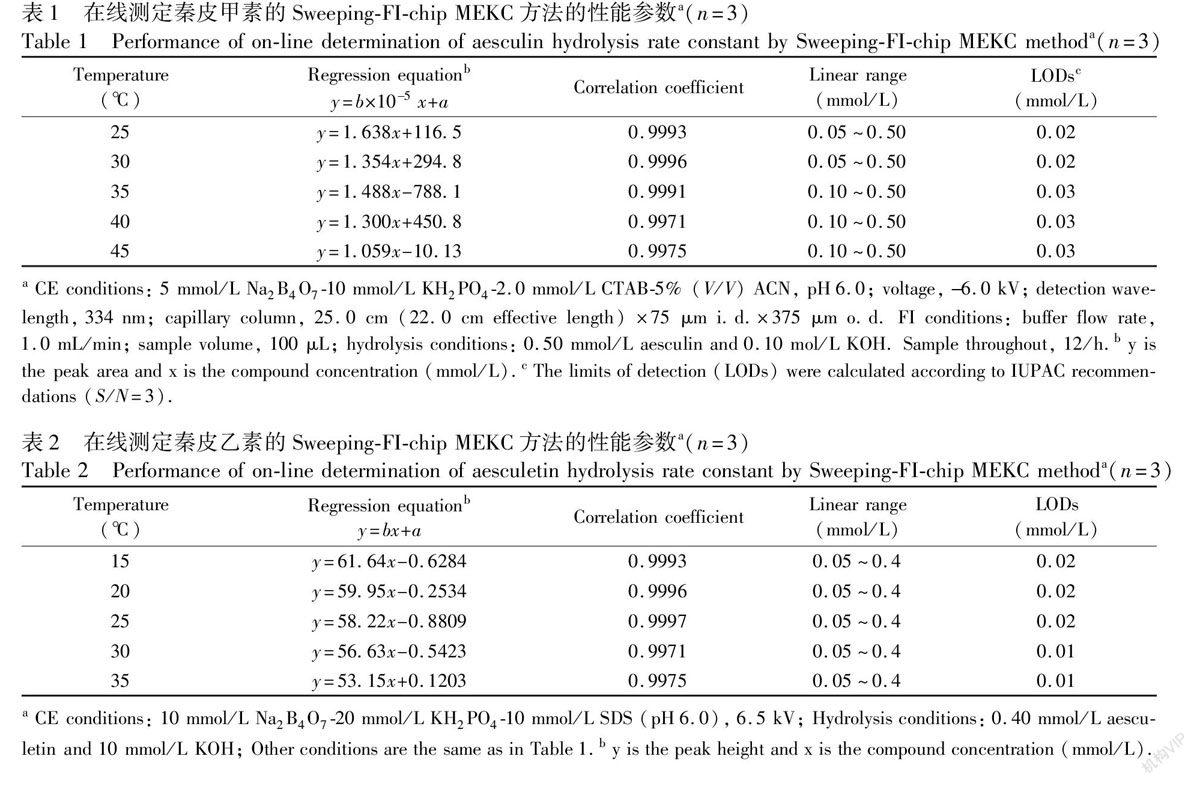
3.2 在線SweepingFIchip MEKC方法的性能
為了準確測定秦皮甲素和秦皮乙素水解反應速率常數,要求用于定量分析二者濃度的方法必須具有高準確度和精密度。為此,我們通過實驗對本文所建立的在線SweepingFIchip MEKC方法的性能進行了考察。在最優條件下,以每5 min進一次樣(12次/h)的頻率連續進樣、分離和測定,結果見表1和表2。由表可見,各個水解反應條件下的反應物的峰面積或峰高與其濃度的回歸方程的線性相關系數均大于0.997,而且秦皮甲素和秦皮乙素兩者的峰面積相對標準偏差分別為1.4%和1.1%, 峰高相對標準偏差分別為1.2%和0.9%, 遷移時間的相對標準偏差分別為0.4%和0.6%。結果表明,本方法可以準確測定秦皮甲素和秦皮乙素的濃度。
上述結果表明,所建立的方法靈敏度高、重現性好,完全滿足在線研究秦皮甲素和秦皮乙素水解動力學的要求。
3.3 秦皮甲素水解速率常數的在線測定
秦皮甲素在30℃時水解反應后所得溶液的電泳譜圖如圖3所示。在秦皮甲素峰的前面有3個未知化合物峰。隨水解時間的增加,其中一個峰的峰高變化不規則,另外兩峰的峰高增加,同時秦皮甲素的峰降低。這些現象表明,3個未知化合物是秦皮甲素的水解產物或水解中間體。有趣的是,這個結果與以往離線用磷酸溶液終止秦皮甲素水解反應后測定其動力學參數的結果不同[11]。這是因為離線測定給出的是秦皮甲素水解平衡狀態的信息,而用SweepingFIchip MEKC在線測定時,秦皮甲素在堿過量條件下進行水解,如前所述,當堿過量時,香豆素結構中的內酯結構很容易解離,水解進行完全,過量的堿一旦被中和(用磷酸溶液終止水解反應),則將重新形成內酯結構,直到達到平衡點,這說明在線測定給出的是秦皮甲素水解過程中的信息。
1, 秦皮甲素; 2~4, 秦皮甲素的水解產物。
Conditions are the same as in Table 1. 1, aesculin; 2-4, the aesculin hydrolyzates
在上述條件下也測定了秦皮甲素在25℃, 35℃, 40℃及45℃的水解速率常數。實驗結果表明,水解速率隨溫度的升高迅速增大。25℃時,秦皮甲素在2 h內水解完全,當溫度升高到45℃時,水解時間縮短為0.5 h。
秦皮甲素在一定的反應時間(t)的瞬時濃度(c)通過如下方法獲得:準確地記錄從反應開始到每次注射時間t,而使用校準曲線計算得到c。以logc對t作圖在各個溫度下均得到一條直線,說明秦皮甲素水解類似一級反應。由于水在水解過程中是大量的,其濃度可認為是常數。因此,水解速率常數k可以用一級動力學方程(1)計算:
3.4 秦皮乙素水解速率常數的在線測定
當用同樣的方法測定秦皮乙素水解速率常數時的電泳譜圖見圖4。由圖可見,由于KOH濃度很大(0.1 mol/L),導致秦皮乙素劇烈水解,25℃時水解30 min即反應完全。要測定秦皮乙素水解速率常數,至少需要5次精確進樣分析,實驗中FIchip CE的采樣頻率為12次/h,25℃時勉強可以通過5次進樣測定秦皮乙素水解反應速率常數,而在30℃, 35℃, 40℃和45℃時則無法測定。考慮到秦皮乙素類藥物一般在室溫和酸堿性溫和的條件下生產或存放,因此測定了1535℃、10 mmol/L KOH條件下秦皮乙素水解反應速率常數。結果見表4。由Arrhenius方程計算的活化能為31.55 kJ/mol。
4 結 論
建立了在線測定秦皮甲素和秦皮乙素水解速率常數的sweeptingFIchip MEKC的方法。本方法不需要終止反應,30 min內可以測定至少5個點的數據,滿足了測定水解反應速率常數對分析速度的要求。與傳統的離線測定化學反應常數的方法相比,本方法簡單、快速、準確。
References
1 Kaneko T, Tahara S, Takabayashi F, Harada N. Free Radical Res., 2004, 38(8): 839-846
2 Wang C J, Hsieh Y J, Chu C Y, Lin Y L, Tseng T H. Cancer Lett., 2002, 183(2): 163-168
3 Biyani M, Nishigaki K. Electrophoresis, 2001, 22(1): 23-28
4 Wang J, Chatrathi M P, Tian B M, Polsky R. Anal. Chem., 2000, 72(11): 2514-2518
5 Kunkel A, Günter S, Wtzig H. Electrophoresis, 1997, 18(10): 1882-1889
6 Kunkel A, Günter S, Wtzig H. J. Chromatogr. A, 1997, 768(1): 125-133
7 Zhang L, Tong P, Chen G N. J. Chromatogr. A, 2005, 1098(1-2): 194-198
8 ZHANG LingYi, REN Jun, PENG Li, LIU Fan, ZHANG WeiBing. Chinese J. Anal. Chem., 2015, 43(10): 1545-1550
張凌怡, 任 俊, 彭 麗, 劉 翻, 張維冰. 分析化學, 2015, 43(10): 1545-1550
9 Chen Y F, Xu L L, Zhao W W, Guo L P, Yang L. Anal. Chem., 2012, 84(6): 2961-2967
10 Kanoatov M, Galievsky V A, Krylova S M, Cherney L T, Jankowski H K, Krylov S N. Anal. Chem., 2015, 87(5): 3099-3106
11 Chen Q H, Hou S X, Zheng J, Bi Y Q, Li Y B, Yang X J, Cai Z, Song X R. J. Chromatogr. B, 2007, 858(12): 199-204
12 Dodge F D. In the Meeting at New Orleans, 1915: 446-448
13 Kuban P, Engstrm A, Olsson J C, Thorsén G, Tryzell R, Karlberg B. Anal. Chim. Acta, 1997, 337(2): 117-124
14 Fang Z L, Liu Z S, Shen Q. Anal. Chim. Acta, 1997, 346(2): 135-143
15 Chen H L, Wang K T, Pu Q S, Chen X G, Hu Z D. Electrophoresis, 2002, 23(17): 2865-2871
16 Cheng Y Q, Fan L Y, Chen H L, Chen X G, Hu Z D. J. Chromatogr. A, 2005, 1072(2): 259-265
17 Fan L Y, Cheng Y Q, Li Y Q, Chen H L, Chen X G, Hu Z D. Electrophoresis, 2005, 26(22): 4345-4354
18 Liu X M, Zhang J S, Chen X G. J. Chromatogr. B, 2007, 852(12): 325-332
19 Liu X M, Liu L H, Chen H L, Chen X G. J. Pharm. Biomed. Anal., 2007, 43(5): 1700-1705
20 Liu L H, Fan L Y, Chen H L, Chen X G, Hu Z D. Electrophoresis, 2005, 26(15): 2999-3006
21 Pan Z W, Chen X G, Hu Z D. Biomed. Chromatogr., 2004, 18(8): 581-588
22 Zhu H D, Lü W J, Li H H, Ma Y H, Hu S Q, Chen H L, Chen X G. Analyst, 2011, 136: 1322-1328
23 Hartwell S K, Kehling A, Lapanantnoppakhun S, Grudpan K. Anal. Lett., 2013, 46(11): 1640-1671
24 Clavijo S, Avivar J, Suárez R, Cerd V. TrAC, Trends Anal. Chem., 2015, 67: 26-33
25 Palmer J, Munro N J, Landers J P. Anal. Chem., 1999, 71(9): 1679-1687
26 Quirino J P, Terabe S. Science, 1998, 282(5388): 465-468
27 Quirino J P, Terabe S. Anal. Chem., 1999, 71(8): 1638-1644
28 Quirino J P, Terabe S, Bocek P. Anal. Chem., 2000, 72(8): 1934-1940
