多重富集定量PCR(ME-qPCR)同時檢測4種食源性病原弧菌
2016-12-09 08:36:09汪天杰周廣彪林春貴吳希陽
中國農業科學 2016年23期
魏 霜,汪天杰,龍 陽,周廣彪,林春貴,黃 帥,吳希陽
?
多重富集定量PCR(ME-qPCR)同時檢測4種食源性病原弧菌
魏 霜1,2,汪天杰3,龍 陽4,周廣彪1,林春貴1,黃 帥1,吳希陽2
(1汕頭出入境檢驗檢疫局,廣東汕頭 515041;2暨南大學理工學院食品科學與工程系,廣州 510632;3廣州出入境檢驗檢疫局,廣州 510632;4湛江出入境檢驗檢疫局,廣東湛江 524022)
【目的】溶藻弧菌、副溶血弧菌、創傷弧菌、霍亂弧菌是4種重要的食源性病原弧菌,能夠造成人類多種疾病,建立同時檢測這4種食源性病原弧菌的檢測方法是保障食品安全的基礎。本研究旨在建立同時檢測溶藻弧菌、副溶血弧菌、創傷弧菌、霍亂弧菌的多重富集定量PCR(multiplex enrichment quantitative PCR,ME-qPCR)方法,并含擴增內標用于指示PCR反應的假陰性,創新一種高通量、高靈敏度、具備定量能力的檢測方法,為這4種食源性病原弧菌的檢測提供新方法。【方法】以細菌為擴增內標靶序列設計引物,并針對副溶血弧菌的、溶藻弧菌的、霍亂弧菌的、創傷弧菌的,分別設計內、外2對特異性引物,首先將所有內、外引物混合,進行一個循環數較少(10—20 cycles)的高通量多重富集PCR將靶基因富集出來,由于循環數較少,各個基因得到均勻的擴增,每個基因均有4種可能的產物,每1種產物均能作為第二輪巢氏熒光定量PCR的模板,這增加了靶基因從模板中被富集出來的概率,然后將產物稀釋后作為模板,利用內引物分別進行巢式熒光定量PCR檢測各個基因,最后根據擴增曲線和熔融曲線分析結果。通過設置不同的第一輪多重富集PCR循環數(10、15和20個循環),優化ME-qPCR第一輪循環數。采用14株標準菌株的基因組DNA作為模板,評價ME-qPCR的特異性。并以4種弧菌基因組DNA混合物梯度稀釋的樣品(100、10、1、0.1、0.01和0.001 ng·μL-1)作為模板,對建立的ME-qPCR進行靈敏度和定量能力的評價。并將該方法應用于已分離到的69株疑似弧菌菌落的鑒定中,與傳統生理生化鑒定結果進行對比。【結果】優化后,ME-qPCR的第一輪循環數確定為15,特異性評價結果顯示該方法特異性強,靈敏度達0.001 ng,高于普通熒光定量PCR約1個數量級,并能有效指示PCR反應的假陰性,且擁有與普通熒光定量PCR相同的定量能力,擴增效率和R符合定量的要求;將建立的ME-qPCR方法應用于69株疑似弧菌菌株的鑒定,24個綠色菌落為副溶血弧菌,22個黃色菌落為溶藻弧菌,1個黃色菌落為創傷弧菌,沒有檢出霍亂弧菌,其結果與生理生化鑒定結果一致。【結論】該方法能夠定量、快速準確地檢測副溶血弧菌、溶藻弧菌、霍亂弧菌和創傷弧菌這4種食源性病原弧菌,靈敏度高,并能有效指示PCR反應的假陰性,結果無需凝膠電泳,適用于食品中4種常見病原弧菌的快速篩檢。
霍亂弧菌;創傷弧菌;副溶血弧菌;溶藻弧菌;擴增內標;多重富集定量PCR
0 引言
【研究意義】弧菌屬()為革蘭氏陰性菌,嗜鹽,廣泛存在于海水中,是引起魚、蝦等水產品疾病的重要病原菌[1-2]。其中,副溶血弧菌、溶藻弧菌、霍亂弧菌和創傷弧菌同時也是重要的食源性病原弧菌[3-4],對人類具有較強的致病性,例如霍亂弧菌能引起人腹瀉、嘔吐等病癥,曾經在世界上多次爆發;溶藻弧菌也早在1979年就有報道稱其對人類的致病性,能夠引起中耳炎等疾病[5],而這些弧菌常常存在于人類食用的海產品中,對海產品中這些弧菌的檢測、監測具有重要意義。【前人研究進展】弧菌的檢測主要包括生理生化鑒定、PCR法等,生理生化鑒定方法周期長,不能滿足快速檢測的要求;而以PCR為基礎的方法由于其高特異性、高靈敏度、快速等特點得到了廣泛的應用。近年來,國內外用于弧菌快速檢測的方法有PCR[6-7]、巢氏PCR[8-9]、多重PCR[10-11]、熒光定量PCR[12-13]、多重熒光定量PCR[14-15]等。多重PCR具有高通量的特點,但由于引物之間的競爭,其檢測通量受到了限制,而且結果需要進一步凝膠電泳分析,費時費力;巢氏PCR擁有高靈敏度的特點,但在其檢測通量方面無法與多重PCR相比;熒光定量PCR因無需凝膠電泳,得到了最為廣泛的應用,但多重熒光定量PCR由于引物探針之間的競爭和儀器熒光檢測通道的限制,難以實現高通量檢測。因此,需要建立一種基于熒光定量PCR的高通量、高靈敏、具備定量能力的檢測方法。【本研究切入點】本研究以4種重要食源性病原弧菌(副溶血弧菌、霍亂弧菌、創傷弧菌、溶藻弧菌)為靶目標,提出一種多重富集定量PCR(multiplex enrichment quantitative PCR, ME-qPCR)方法,改變現有方法存在的缺陷,真正實現高通量、高靈敏度、定量檢測。【擬解決的關鍵問題】本研究擬針對常見的4種食源性病原弧菌——副溶血弧菌的、溶藻弧菌的、霍亂弧菌的和創傷弧菌的,分別設計特異性內、外引物,并針對細菌保守區設計通用引物作為擴增內標,建立同時能檢測4種食源性病原弧菌的ME-qPCR體系,同時能夠指示PCR反應的假陰性,結果無需凝膠電泳,真正達到定量、高通量、快速、準確的檢測要求。
1 材料與方法
試驗于2015年9月至2016年3月在汕頭出入境檢驗檢疫局檢驗檢疫技術中心進行。
1.1 材料
河流弧菌、擬態弧菌、鰻弧菌、哈維氏弧菌、梅氏弧菌保存于汕頭出入境檢驗檢疫局檢驗檢疫技術中心動物檢疫實驗室。副溶血弧菌(ATCC17802)、溶藻弧菌(ATCC17749)、霍亂弧菌(ATCC14035)、創傷弧菌(ATCC27562)、大腸桿菌(ATCC8739)、單增李斯特菌(ATCC19111)、金黃色葡萄球菌(ATCC43300)、枯草芽孢桿菌(ATCC6633)、傷寒沙門氏菌(ATCC14028)保存于暨南大學食品科學與工程系。
1.2 細菌培養及基因組DNA的提取
將弧菌單菌落接種于含3% NaCl的胰蛋白胨大豆肉湯(tryptic soy broth,TSB)培養基中,將非弧菌單菌落接種于腦心浸液(brian heart infusion,BHI)液體培養基中,37℃下振蕩培養過夜。采用試劑盒法(TIANamp Bacteria DNA kit,TIANGEN)提取細菌基因組DNA,提取后取1 μL用微量紫外分光光度計(ND-1000,NanoDrop)測定提取的基因組DNA的濃度和純度,基因組DNA保存于-20℃待用。
1.3 引物的設計與合成
根據副溶血弧菌(GenBank ID:AF326572.1)、溶藻弧菌(GenBank ID:AF007288)、霍亂弧菌(GenBank ID:X51948.1)和創傷弧菌(GenBank ID:AF376027.1)序列,以及細菌的保守序列,利用Primer Premier V5.0和Oligo V6.22軟件進行引物的設計和篩選,并保證內、外引物產物大小符合熒光定量PCR的要求(小于200 bp)。在參考了大量文獻已報道的引物基礎上篩選出特異性外引物,在外引物產物片段序列內設計內引物,副溶血弧菌、溶藻弧菌和創傷弧菌的內引物設計較困難,故保留外引物中特異性較強的那一條,僅設計一條內引物,形成3對半巢式引物組,16S rRNA基因作為IAC的靶基因引物參考了已報道的文獻,但內引物設計細菌通用引物難度較大,內外引物均用同一對引物。上述所有 引物由生工生物工程(上海)股份有限公司合成(表1)。

表1 各基因的內外引物序列
1.4 ME-qPCR體系
多重富集定量PCR(Multiplex multiplex enrichment quantitative PCR,ME-qPCR)方法針對每一個靶基因序列均設計內、外兩對引物,內外引物產物大小符合基于SYBR Green I的熒光定量PCR的要求。首先,將所有內、外引物全部混合,進行一個循環數較少(10—20 cycles)的高通量多重富集PCR,然后將PCR產物用ddH2O稀釋作為模板,運用內引物進行第二輪巢式熒光定量PCR,結果根據擴增曲線和熔融曲線分析。在第一輪高通量多重PCR中,每個靶基因均有4種可能的產物類型,這4種產物類型均能作為第二輪巢式熒光定量PCR的模板,這意味著4條引物對應的4種組合只要有1種能夠成功擴增,整個反應就能進行,這相當于將靶基因從模板中富集(enrichment)出來,這增加了本方法的成功率和穩定性。同時,由于第一輪循環數較少,引物之間的競爭較弱,各個基因均能得到均勻擴增(圖1)。具體操作步驟如下:
第一輪多重富集PCR采用多重PCR反應試劑盒(Multiplex PCR Assay Kit,TaKaRa),反應體系為:Mix2溶液25 μL,Mix1溶液0.25 μL,各基因的內、外引物混合物(各個引物終濃度均為0.1 μmol·L-1)和DNA模板各2 μL,用ddH2O調整體積至50 μL。反應條件為:94℃ 1 min;94℃ 30 s,60℃ 60 s,72℃ 60 s,10—20個循環;72℃ 5 min(Mastercycler ep,Eppendorf)。
將第一輪多重富集PCR產物用ddH2O稀釋100倍作為第二輪巢式熒光定量PCR的模板,使用SYBR熒光定量PCR反應試劑盒(SYBR Premix Ex Taq,TaKaRa),每孔體系為:SYBR溶液12.5 μL、第一輪PCR產物稀釋物1 μL、各基因的內引物(引物終濃度均為0.4 μmol·L-1),用ddH2O調整體積至25 μL。反應條件為95℃ 30 s;95℃ 5 s,60℃ 10 s,72℃ 10 s,35個循環,每個循環在72℃結束后檢測熒光信號;反應結束后進行熔融曲線分析:65—95℃,每0.5℃掃描熒光一次(CFX96,Bio-rad)。
1.5 ME-qPCR體系特異性的評價
利用1.1中的14株菌株,按照1.2的的方法提取基因組DNA,并按照1.4步驟進行檢測,對結果進行分析,評價ME-qPCR體系的特異性。
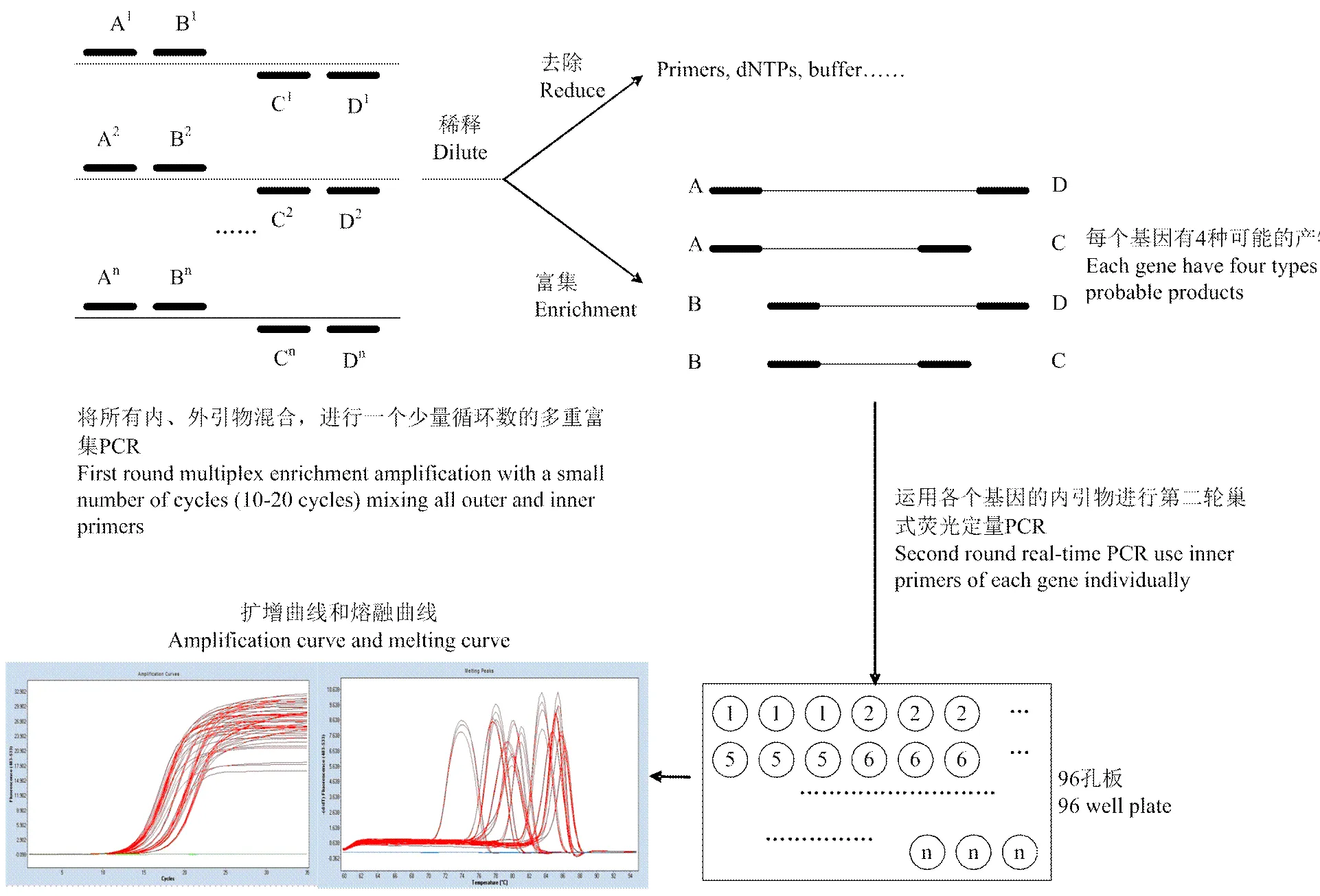
圖1 ME-qPCR的原理
1.6 ME-qPCR體系的定量能力和靈敏度評價
按照1.2的方法分別提取溶藻弧菌、副溶血弧菌、霍亂弧菌和創傷弧菌的基因組DNA,采用10倍梯度稀釋,分別制備成濃度分別為100、10、1、0.1、0.01和0.001 ng·μL-1的樣品,對上述樣品分別采用ME-qPCR和普通熒光定量PCR進行檢測,普通熒光定量PCR直接采用內引物組,對比兩種方法的定量能力和靈敏度,評價ME-qPCR的定量能力和靈敏度。
1.7 ME-qPCR體系檢測實際樣品
采用本課題組前期分離的69株疑似弧菌菌株[20],利用已建立的ME-qPCR體系對上述弧菌菌落進行鑒定,同時對這些疑似菌落進行生理生化鑒定,對比兩種方法。
2 結果
2.1 ME-qPCR第一輪反應循環數的優化
第一輪多重富集PCR的循環數是ME-qPCR的關鍵參數。將4種弧菌的基因組DNA混合作為模板,進行第一輪多重富集循環數分別為10、15和20的ME-qPCR,并直接運用內引物對上述模板進行普通熒光定量PCR,對比在不同第一輪反應循環數下ME-qPCR與普通熒光定量PCR的差別。結果如表2所示,當第一輪反應循環數為10時,其Ct值大于普通熒光定量PCR,說明其靈敏度低于普通熒光定量PCR;當第一輪反應循環數為15和20時,其Ct值均小于普通熒光定量PCR。但理論上第一輪反應循環數越小,引物之間的競爭越少,PCR反應偏好性越弱。綜合考慮,確定第一輪反應循環數為15。
2.2 ME-qPCR體系的建立
在確定了ME-qPCR第一輪反應的循環數后,混合溶藻弧菌、副溶血弧菌、創傷弧菌和霍亂弧菌的基因組DNA為模板,建立了ME-qPCR體系用于4種食源性病原弧菌的檢測,同時在體系中添加IAC能指示PCR反應的假陰性。結果如圖2所示,5個基因均得到擴增,每個基因3個復孔的重復性好,熔融曲線結果表明產物單一,沒有非特異性擴增產物的出現。
2.3 ME-qPCR體系特異性評價
利用14株菌對ME-qPCR體系的特異性進行了評價。結果如圖3所示,14種菌的IAC均表現為陽性擴增,說明反應不存在假陰性,4種目標菌順利檢出,其余非目標菌均為陰性結果,表明建立的ME-qPCR體系特異性強。
2.4 ME-qPCR體系定量能力及靈敏度的評價
混合4種食源性病原弧菌的基因組DNA并稀釋成一系列濃度梯度,同時進行ME-qPCR和普通熒光定量PCR,對比兩種方法的定量能力和靈敏度。結果如表3所示,ME-qPCR的靈敏度高于普通熒光定量PCR約1個數量級,2種方法的Ct值線性關系好,2均大于0.99,PCR擴增效率符合熒光定量PCR的要求,這說明ME-qPCR擁有和普通熒光定量PCR相同的定量能力。
2.5 ME-qPCR體系檢測實際樣品
利用建立好的ME-qPCR體系對課題組前期分離的69個疑似弧菌菌落進行PCR鑒定,結果與前期的API生理生化試驗結果一致[20]。
3 討論
引物之間的競爭一直是以PCR為基礎的高通量檢測技術的瓶頸,傳統多重PCR通過設計引物時降低理論上引物之間形成二級結構的可能性來提高多重PCR的成功率,而且同時要考慮不同靶基因的產物片段大小,這兩方面加在一起大大增加了引物的設計難度,因此有學者開發了多重PCR引物設計系統[21],這一定程度上提高了構建多重PCR體系的成功率。另外,還有學者將多重PCR與毛細管電泳技術[22],或者改進引物的設計方法,例如DPO(dual priming oligonucleotide)引物[23]、通用引物多重PCR(universal primer multiplex PCR,UP-M-PCR)[24]等,從而提高了多重PCR的檢測通量。但多重PCR的檢測通量依然受到多方面限制,而且多重PCR需要凝膠電泳或毛細管電泳分析,增加了檢測所需的時間。本研究建立的多重富集定量PCR(multiplex enrichment quantitative PCR,ME-qPCR)體系分2步反應,首先進行一輪高通量的多重富集PCR,然后將產物稀釋作為模板分別進行第二輪熒光定量PCR。由于第一輪反應的循環數較少,引物之間的競爭減少,各個基因能得到均勻擴增,這與多重串聯式PCR(multiplex tandem PCR,MT-PCR)的原理一致[25]。另外,將內、外引物同時添加進第一輪反應中,會產生4種可能的產物,這4種可能的產物均能夠作為第二輪巢式熒光定量PCR的模板,這相當于增加了模板被富集出來的概率,從而提高了反應的成功率,這與擴增子拯救多重PCR(amplicon rescue multiplex PCR,Arm-PCR)的原理一致[26]。本研究建立的ME-qPCR體系既提高了檢測的靈敏度,又能保證整個體系的定量能力,同時提高了檢測通量,結果無需凝膠電泳,適合實驗室對這4種常見食源性病原菌的檢測。

表2 ME-qPCR第一輪循環數的優化
多重富集定量PCR(Multiplex enrichment quantitative PCR,ME-qPCR);熒光定量PCR(Real-time quantitative PCR,RT-qPCR)。下同 The same as below
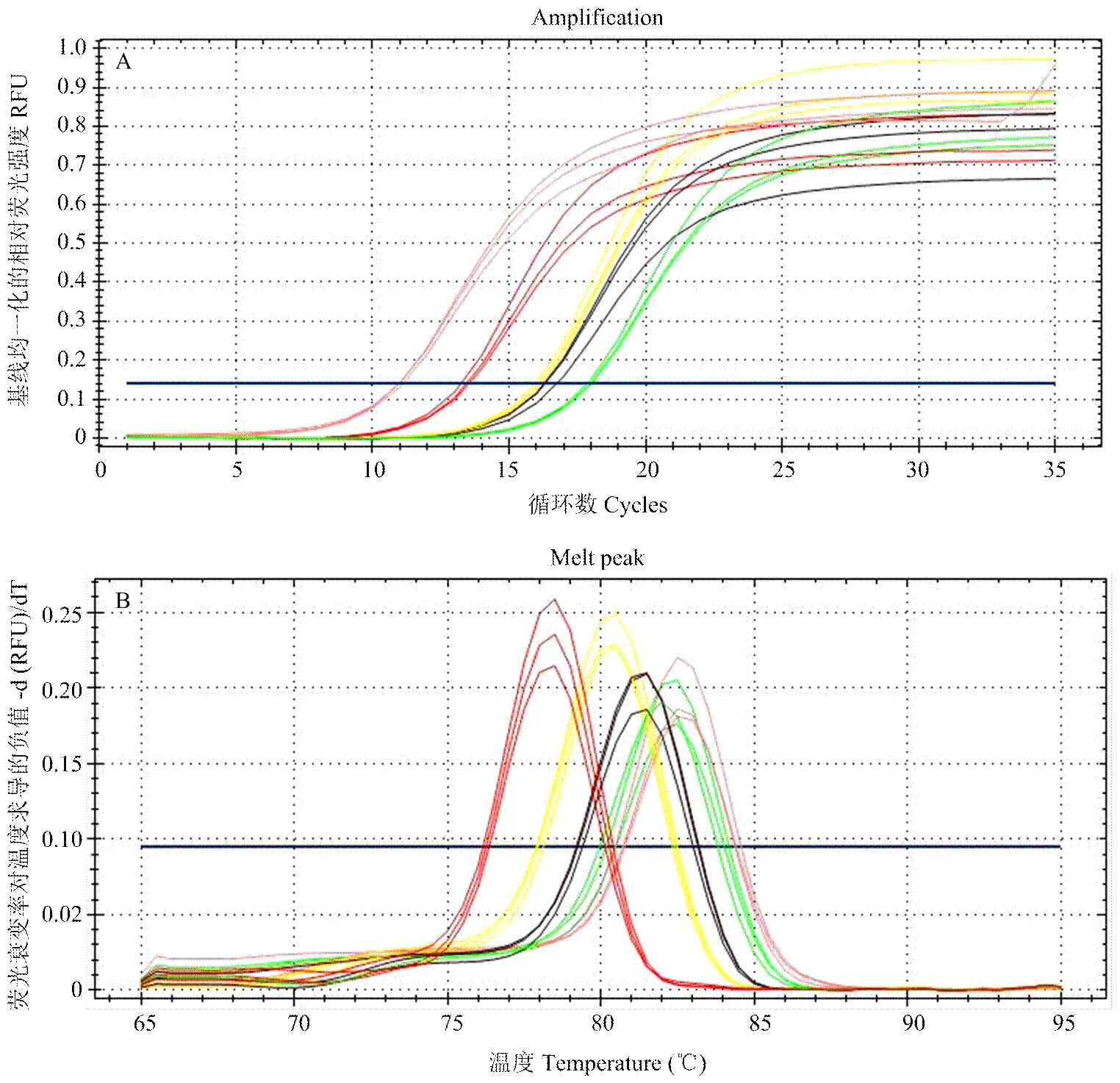
A圖為擴增曲線;B圖為熔融曲線 Fig A is amplification curve; Fig B is melting curve
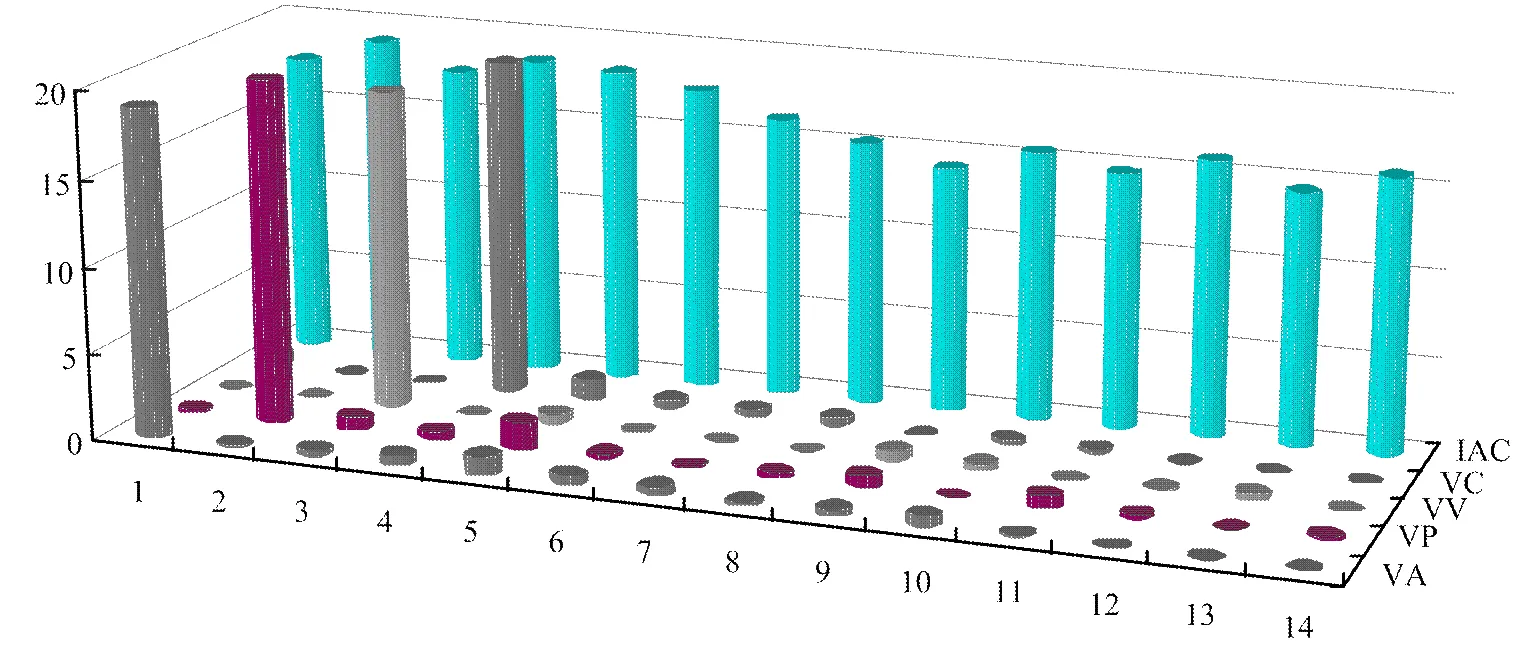
Z軸坐標代表35-Ct值;X軸坐標VA、VP、VV、VC、IAC分別代表溶藻弧菌、副溶血弧菌、創傷弧菌、霍亂弧菌的特異性引物和擴增內標;Y軸坐標1—14分別代表溶藻弧菌、副溶血弧菌、創傷弧菌、霍亂弧菌、擬態弧菌、河流弧菌、哈維氏弧菌、梅氏弧菌、鰻弧菌、大腸桿菌、單增李斯特菌、金黃色葡萄球菌、枯草芽孢桿菌、傷寒沙門氏菌
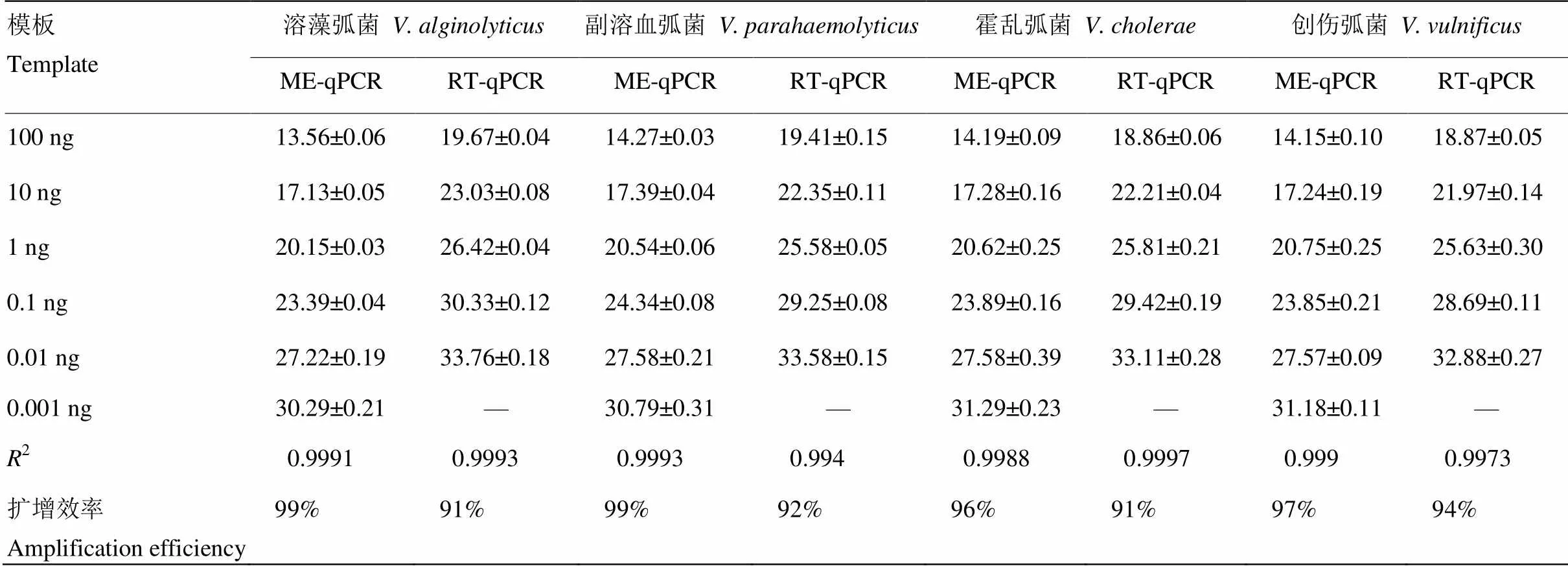
表3 多重富集定量PCR(ME-qPCR)與實時定量PCR(RT-qPCR)靈敏度及定量能力對比
—:未檢出 Not detected
靈敏度是基于PCR檢測方法的一個重要方面,熒光定量PCR的靈敏度一般高于普通PCR,而巢氏熒光定量PCR靈敏度又高于熒光定量PCR。COSTA等[27]建立了一種單管巢氏熒光定量PCR,高于普通熒光定量PCR法1個數量級。本研究建立的ME-qPCR方法,靈敏度高達0.001 ng,同樣高于普通熒光定量PCR法1個數量級,這是由于ME-qPCR的第一輪進行了一次高通量多重富集PCR,然后將產物作為第二輪巢氏熒光定量PCR,這與巢氏PCR的效果相當,因而提高了該方法的靈敏度。
在實際檢測過程中,樣品中存在PCR抑制因子,或者儀器的故障等因素,都會導致PCR結果假陰性的出現[28],影響結果的準確性。目前,在PCR檢測體系中添加擴增內標(internal amplification control,IAC)是指示是否存在PCR反應假陰性的重要方法[29]。Nordstrom等[30]針對副溶血弧菌、和這3類溶血素基因,建立了含擴增內標的多重熒光定量PCR,在檢測的同時能指示PCR反應的假陰性,提高了檢測的準確性。魏霜等[20]針對溶藻弧菌、副溶血弧菌、創傷弧菌和霍亂弧菌,建立了添加擴增內標的多重PCR。本研究建立的ME-qPCR體系內添加了以為靶基因的擴增內標,能有效指示PCR反應的假陰性,提高了檢測的準確性。
4 結論
本研究建立了一套多重富集定量PCR用于同時檢測溶藻弧菌、副溶血弧菌、創傷弧菌和霍亂弧菌,并能有效指示PCR反應的假陰性,該方法特異性強,比普通熒光定量PCR靈敏度高1個數量級,而且擁有與普通熒光定量PCR相同的定量能力,結果無需凝膠電泳分析,適用于食品中4種常見病原弧菌的快速篩檢。由于該方法同時兼顧了高通量、高靈敏度、定量等方面的優勢,為某些痕量樣品的分析如石蠟包埋樣品各基因表達量的分析、深加工食品轉基因成分檢測等提供了一種新方法;同時,由于本方法是基于染料法的熒光定量PCR,也可將該方法與高分辨率溶解曲線(high resolution melting,HRM)技術相結合,用于SNP位點的檢測,適合于某些大量樣品、多位點的分析。
References
[1] Toranzo A E, Magarinos B, Romalde J L. A review of the main bacterial fish diseases in mariculture systems., 2005, 246: 37-61.
[2] Baffone W, Citterio B, Vittoria E, Casaroli A, Pianetti A, Campana R, Bruscolini F. Determination of several potential virulence factors inspp. Isolated from sea water., 2001, 18(5): 479-488.
[3] Zhang X H, Austin B. Haemolysins inspecies., 2005, 98(5): 1011-1019.
[4] SU Y C, LIU C C.: A concern of seafood safety., 2007, 24(6): 549-558.
[5] SCHMIDT U, CHMEL H, COBBS C.infections in humans., 1979, 10(5): 666-668.
[6] 韓一凡, 莫照蘭, 李杰, 茅云翔, 肖鵬, 汪笑宇, 楊官品. 溶藻弧菌的PCR快速檢測方法. 中國海洋大學學報(自然科學版), 2009, 39(6): 1237-1240.
HAN Y F, MO Z L, Li J, MAO Y X, XIAO P, WANG X Y, YANG G P. The detection ofby PCR assay., 2009, 39(6): 1237-1240. (in Chinese)
[7] KIM Y B, OKUDA J, MATSUMOTO C, TAKAHASHI N, HASHIMOTO S, NISHIBUCHI M. Identification ofstrains at the species level by PCR targeted to the toxR gene., 1999, 37(4): 1173-1177.
[8] ARIAS C R, GARAY E, AZNAR R. Nested PCR method for rapid and sensitive detection ofin fish, sediments, and water., 1995, 61(9): 3476-3478.
[9] LEE S E, KIM S Y, KIM S J, KIM H S, SHIN J H, CHOI S H, CHUNG S S, RHEE J H. Direct identification ofin clinical specimens by nested PCR., 1998, 36(10): 2887-2892.
[10] NEOGI S B, CHOWDHURY N, AASKURA M, HINENOYA A, HALDAR S, SAIDI S M, KOGURE K, LARA R J, YAMASAKI S. A highly sensitive and specific multiplex PCR assay for simultaneous detection of,and., 2010, 51(3): 293-300.
[11] HALDAR S, NEOGI S B, KOGURE K, CHATTERJEE S, CHOWDHURY N, HINENOYA A, ASAKURA M, YAMASAKI S. Development of a haemolysin gene-based multiplex PCR for simultaneous detection of,and., 2010, 50(2): 146-152.
[12] BLACKSTONE G M, NORDSTROM J L, VICKERY M C L, BOWEN M D, MEYER R F, DEPAOLA A. Detection of pathogenicin oyster enrichments by real time PCR., 2003, 53(2): 149-155.
[13] MCCLEARY S, HENSHILWOOD K. Novel quantitative TaqMan? MGB real-time PCR for sensitive detection ofin., 2015, 114(3): 239-248.
[14] GUBALA A J. Multiplex real-time PCR detection of., 2006, 65(2): 278-293.
[15] HE P, CHEN Z, LOU J, WANG H, YAN Y, CHEN L, GAO W. Multiplex real-time PCR assay for detection of pathogenicstrains., 2014, 28(5): 246-250.
[16] PANICKER G, MYERS M L, BEJ A K. Rapid detection ofin shellfish and gulf of Mexico water by real-time PCR., 2004, 70(1): 498-507.
[17] PINTO A D, CICCARESE G, TANTILLO G, CATALANO D, FORTE V T. A collagenase-targeted multiplex PCR assay for identification of,, and., 2005, 68(1): 150-153.
[18] ZHOU S, HOU Z, LI N, QIN Q. Development of a SYBR Green I real-time PCR for quantitative ofin seawater and seafood., 2007, 103(5): 1897-1906.
[19] NANDI B, NANDY R K, MUKHOPADHYAY S, NAIR G B, SHIMADA T, GHOSE A C. Rapid method for species-specific identification ofusing primers targeted to the gene of outer membrane protein ompW., 2000, 38(11): 4145-4151.
[20] 魏霜, 冼鈺茵, 趙暉, 吳希陽. 多重PCR檢測四種食源性病原弧菌. 中國農業科學, 2013, 46(8): 1682-1686.
WEI S, XIAN Y Y, ZHAO H, WU X Y. Multiplex PCR assays for the detection of,,and., 2013, 46(8): 1682-1686. (in Chinese)
[21] 申志勇. PCR引物特異性評估體系及多重PCR引物設計系統的構建與應用[D]. 北京: 中國人民解放軍軍事醫學科學院, 2009.
SHEN Z Y. The construction and application of the PCR primer specificity evaluating system and multiplex PCR primer design system [D]. Beijing: Academy of Military Medical Sciences, 2009. (in Chinese)
[22] YAN C, SUN H, XUE G, ZHAO H, WANG L, FENG Y, LI S. A single-tube multiple-locus variable-number tandem-repeat analysis ofclinical specimens by use of multiplex PCR-capillary electrophoresis., 2014, 52(12): 4168-4171.
[23] XU Y G, LIU Z M, GUAN X T, CUI L C, LI S L. Dual priming oligonucleotide (DPO)-based multiplex PCR assay for specific detection of four diarrhoeagenicin food., 2015, 61(2): 146-152.
[24] XU W, ZHAI Z, HUANG K, ZHANG N, YUAN Y, SHANG Y, LUO Y. A novel universal primer-multiplex-PCR method with sequencing gel electrophoresis analysis., 2012, 7(1): e22900.
[25] STANLEY K K, SZEWCZUK E. Multiplexed tandem PCR: gene profiling from small amounts of RNA using SYBR Green detection., 2005, 33(20): e180.
[26] 耿偉光, 史成銀, 李晉, 黃倢, 王秀華, 謝國駟. 同步檢測海水養殖動物5種病原菌的擴增子拯救多重 PCR (Arm-PCR) 方法的建立與應用. 農業生物技術學報, 2013, 21(9): 1125-1134.
GENG W G, SHI C Y, LI J, HUANG J, WANG X H, XIE G S. Establishment and application of amplicon rescue multiplex PCR (Arm-PCR) for the simultaneous detection of five major bacterial pathogens from marine aquaculture animals., 2013, 21(9): 1125-1134. (in Chinese)
[27] COSTA J, MAFRA I, KUCHTA T, OLIVEIRA M B P P. Single-tube nested real-time PCR as a new highly sensitive approach to trace hazelnut., 2012, 60(33): 8103-8110.
[28] ABUALSOUD W, RADSTROM P. Purification and characterization of PCR-inhibitory components in blood cells., 2001, 39(2): 485-493.
[29] HOORFAR J, MALOMY B, ABDULMAWJOOD A, COOK N, WAGNER M, FACH P. Practical considerations in design of internal amplification controls for diagnostic PCR assays., 2004, 42(5): 1863-1868.
[30] NORDSTROM J L, VICKERY M C L, BLACKSTONE G M, MURRAY S L, DEPAOLA A. Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenicbacteria in oysters., 2007, 73(18): 5840-5847.
(責任編輯 趙伶俐)
Multiplex Enrichment Quantitative PCR Assays for the Detection of,,and
WEI Shuang1,2, WANG Tian-jie3, LONG Yang4, ZHOU Guang-biao1, LIN Chun-gui1, HUANG Shuai1, WU Xi-yang2
(1Shantou Entry-Exit Inspection and Quarantine Bureau, Shantou 515041, Guangdong;2Department of Food Science and Engineering, College of Science and Engineering, Jinan University, Guangzhou 510632;3Guangzhou Entry-Exit Inspection and Quarantine Bureau, Guangzhou 510632;4Zhanjiang Entry-Exit Inspection and Quarantine Bureau, Zhanjiang 524022, Guangdong)
【Objective】,,andhave been recognized as the important foodborne pathogens causing human disease. It is important to establish a detection method to identify these 4 foodborne pathogenicspecies in order to ensure food safety. To develop a multiplex enrichment quantitative PCR (ME-qPCR) assay that can simultaneously detect,,andin the presence of an internal amplification control (IAC), to make it possible for researchers and technical staff achieve simple detection of these 4 foodborne pathogens. 【Method】 Inner and outer species-specific PCR primers were designed based ongene for,gene for,gene forandgene for,gene of bacteria as IAC primers was used to indicate false-negative results. All inner and outer primers were used in first round multiplex enrichment PCR with a small number of cycles so as to avoid competition between amplicons. Each gene has 4 types of probable products which can be the templates for the next nested real-time PCR. This can enrich the target genes from the genomic DNA template successfully. The reaction product was then diluted and analyzed individual real-time PCRs using inner primers. The results were analysed by amplification curve and melting curve. ME-qPCR method was developed after optimization of the first round multiplex enrichment PCR cycle numbers (10, 15 and 20 cycles). The specificity of ME-qPCR was validated by 14 bacteria standard strains. The sensitivity and quantitative capability of ME-qPCR were tested by using 10-fold serially diluted genomic DNA of 4strains. Strains as templates, and then, the ME-qPCR was used to detect 69 suspiciousstrains and the results were compared with physiological and biochemical experiments. 【Result】Fifteen cycles were determined to use in first round multiplex enrichment PCR in this study finally. The results showed that the ME-qPCR assay was rapid, high-throughput, sensitive and specificand the existence of IAC could successfully eliminate false-negative results. The sensitivity of ME-qPCR was 0.001ng per reaction, about 10 times higher than real-time PCR. The ME-qPCR was validated with 69 suspiciousstrains. The result showed that 27 green bacteria colonies were, 22 yellow bacteria colonies were, 1 yellow bacteria colony was, ande was not detected. The results are consistent with physiological and biochemical experiments. 【Conclusion】 The ME-qPCR assay is specific, stable and reliable for the detection of,,and. It’s sensitivity is high, and can effectively indicate the false negative of PCR reaction, the results without gel electrophoresis, and is suitable for the rapid screening of 4 common pathogenicin food.
;;;; internal amplification control; ME-qPCR
2016-05-03;接受日期:2016-06-27
廣東省科技計劃項目-國際科技合作領域(2015A050502030)、湛江市科技計劃項目(2014C01007)、廣東出入境檢驗檢疫局科技計劃項目(2015GDK27)
魏霜,E-mail:weishuang2008@hotmail.com。通信作者吳希陽,E-mail:tkentwu@jnu.edu.cn
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
