氫溴儲能電池結構的優化和運行條件對電池性能的影響
2016-12-29 08:20:18史繼誠徐洪峰
物理化學學報 2016年12期
關鍵詞:效率
史繼誠 徐洪峰,* 盧 璐 高 俊
(1大連交通大學環境與化學工程學院,遼寧省新能源電池重點實驗室,遼寧大連116028;2黑龍江大慶煉化公司,黑龍江大慶163411)
氫溴儲能電池結構的優化和運行條件對電池性能的影響
史繼誠1徐洪峰1,*盧 璐1高 俊2
(1大連交通大學環境與化學工程學院,遼寧省新能源電池重點實驗室,遼寧大連116028;2黑龍江大慶煉化公司,黑龍江大慶163411)
研究了氫溴電池的電池結構、正極氫溴酸和溴電解質濃度、負極的氫氣壓力、質子交換膜厚度對氫溴電池的性能和電池效率的影響。對氫溴電池結構進行改進,單電池實現了200 mA·cm-2電流密度恒流充放電,電池庫倫效率100%。溴電極電化學反應受濃差極化控制,提高氫溴酸濃度,電池充電性能提高,同時,溴在氫溴酸的溶解度增大,電池放電性能也提高,氫溴酸濃度由0.5 mol·L-1提高至1 mol·L-1,電流密度200 mA·cm-2,電池的能量效率和電壓效率提高27.9%。氫溴電池充電過程,降低電池負極氫出壓力,有利于提高充電性能,但膜透酸嚴重,放電過程中最佳的氫出壓力是維持氫在碳紙憎水催化層的單層吸附,充放電過程氫出壓力均為40.0 kPa,電池的能量效率80.2%。膜厚度與膜電阻極化和膜透酸密切相關,充電過程,膜由50.0 μm降至15.0 μm,膜透酸嚴重,負極電化學活性比表面積下降,電池充電性能降低。膜厚度對放電性能的影響還與電流密度有關,電流密度較低時,膜透酸造成負極電化學比表面積下降居主導地位,50.0 μm Nafion膜放電性能更高;電流密度超過200 mA·cm-2時,膜電阻極化居主導電位,15.0 μm Nafion膜性能更高。采用20.0 μm質子交換膜,在200 mA·cm-2電流密度循環充放電五次,電池的能量效率和電壓效率達到85.3%,庫倫效率100%。
氫溴電池;膜電極;質子交換膜;膜透酸;電池效率
1 引言
節約化石能源,提高化石能源的利用率,開發風能、太陽能、潮汐能等可再生能源,已成為我國可持續發展的重要戰略1。可再生能源發電具有不連續、不穩定的特點,迫切需要建立大規模高效儲能技術,來解決可再生能源發電并網的問題。在現有的化學儲能方式中,液流電池因存儲容量大、安全、循環壽命長而被認為最適宜規模化儲能。目前已對液流電池體系做了許多研究,如Fe-Cr體系2,3、全釩體系4,5、鋅-溴體系6,7、多硫化鈉-溴體系8,9、鐵-釩體系10,11等,但是這類電池仍然存在電池功率密度低、成本高等方面的挑戰,其廣泛應用尚未實現。與以上電池體系相比較,氫溴電池的電-電轉化效率高達80%,電池具有成本低、能量密度和功率密度高等突出優勢,是目前最具競爭力的大規模儲能技術之一12。但是溴和氫溴酸具有高毒性和強腐蝕性,對電極材料密封要求高和實驗條件苛刻。國內對于氫溴儲能電池的研究很少,需要在氫溴電池儲能技術方面有所突破和創新,為氫溴電池的商業化做好技術儲備。
氫溴電池電極反應式如下:
充電:正極 2HBr=Br2+2H++2e
負極 2H++2e=H2
放電:正極 Br2+2H++2e=2HBr
負極 H2=2H++2e (E?=1.09 V)
國外對氫溴電池研究較早,但最近的研究成果較少,Lin等13在氫溴電池中采用Nafion/聚偏二氟乙烯電紡復合膜,摻氮功能化的鉑銥催化劑,以碳納米管為基體的溴電極,通過這些措施,降低了傳統氫溴電池的材料成本,并解決鉑催化劑被HBr和Br2的腐蝕問題。Cho等14認為碳電極氧化為CO2和充電過程中溴化物從正極向負極的穿流,是影響氫溴電池循環性能的關鍵因素。國內在一些電池的研究方面涉及到溴電極,Wang等15采用雙模版法制備了一種高度有序的雙峰介孔材料,材料存在5 nm的介孔和在介孔壁上形成的2 nm孔道,這種結構縮短了傳質距離和傳質阻力,并有利于Br2的吸附,增加了Br2/Br-反應的活性位點。Zhang等16認為鋅溴電池工作電流密度低的原因是溴電極的電化學活性低和電池的內電阻高,其中溴的吸附是溴電極電化學反應的速率控制步驟,如將高比表面積的活性碳負載到膜上,可有效降低電池內阻和提高溴電極電化學反應活性。另外,氫溴電池的電池結構是決定電池是否高效運行的關鍵,H?hne和Starbeck17發明一種氫溴燃料電池結構,電池端板采用石墨,溴電極和氫電極流場為石墨氈,氫電極在石墨上鍍鉑,這種電池結構存在催化活性差、接觸電阻大的缺點,并且解決不了溴的穿流問題。Braff等18報道了無隔膜結構的氫溴層流電池,在200 mW·cm-2功率密度下,電池的電壓效率達到90%,庫倫效率為66%,能量效率為60%,這種無隔膜電池結構,能量效率低于80%,顯然不適合商業化應用。
本文通過氫溴電池結構的優化,在國內首次使電池的能量效率達到了商業化應用的要求,即大于80%,并且研究了多種條件如酸濃度、氫氣壓力和質子交換膜厚度對電池能量效率的影響,更重要的是采用新的電池結構,解決了膜透酸問題,在氫溴電池的商業化應用方面,邁出了堅實的一步。
2 實驗部分
2.1 氫溴電池的工藝裝置和電池結構
目前,氫溴電池用電解質膜主要有兩種,一種是全氟磺酸(Nafion)膜,另一種是納米多孔膜。Nafion膜由于電導率高,結構穩定而廣泛應用于質子交換膜燃料電池和固體聚合物電解質水電解池中19-24。Nafion膜中質子的傳導依賴于水的傳遞,磺酸基團與水結合后將球形面打開提供質子通道,沒有帶電荷的物質(如Br2)也可以從通道穿過25-28。因此,在氫溴電池中使用高濃度的電解液時,Nafion膜不能完全阻止溴離子和溴的滲透而吸附在氫電極上,使鉑催化劑中毒導致氧化還原活性下降。
本文在氫溴電池工藝方面采取了以下措施:一,在氫溴電池的氫氣進出口處各設置一個排酸罐,氫氣經減壓閥首先進入排酸罐,由排酸罐出口進入氫溴電池的負極,負極滲入的酸液由氫氣吹掃排入另一排酸罐。二,適當提高負極側氫氣壓力,減少正負極之間的壓力差,避免膜透酸的同時,起到保護膜電極的作用。三,對催化劑負載膜電極(CCM膜電極)進行改進,負極碳紙擴散層經憎水處理后,負載催化劑。圖1是氫溴電池的工藝裝置,氫溴酸由氫溴酸儲槽經磁力泵打入氫溴電池的進口,電池出口的氫溴酸經冷卻后進入儲槽。
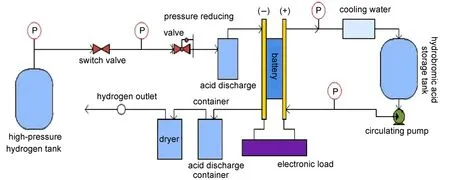
圖1 氫溴電池工藝裝置圖Fig.1 Craftwork device diagram of the hydrogen bromine battery
氫溴電池的第一種結構如圖2所示,Nafion 212膜(厚度50.0 μm,美國杜邦)作為電池隔膜;Nafion膜和聚丙烯腈基碳氈(厚5.0 mm,上海力碩復合材料科技有限公司)均負載Nafion樹脂(美國杜邦)和1 mg·cm-2導電炭黑(BP2000,美國卡博特),構成氫溴電池的正極;Nafion膜的另一側負載Nafion樹脂和0.4 mg·cm-250%Pt/C(日本TANAKA),構成氫溴電池的負極;膜電極面積186 cm2。
氫溴電池的另一種電池結構如圖3所示,在氫溴電池負極的碳紙擴散層上,建立憎水催化層,即負載聚四氟乙烯(PTFE)和0.2 mg·cm-250%Pt/C;Nafion膜負極親水催化層組成為Nafion樹脂和0.2 mg·cm-250%Pt/C;正極的組成同第一種電池結構的正極,膜電極面積186 cm2。
氫溴電池測試的實驗條件如表1所示,氫溴酸(40%(質量分數),天津市北聯精細化學品開發有限公司)稀釋后濃度為0.5 mol·L-1,在電流密度為200 mA·cm-2時,經計算,氫溴酸的最低流量是0.048 L·min-1,受氫溴酸泵出口閥的開度限制,實際氫溴電池入口酸流量為0.183 L·min-1。
2.2 氫溴酸濃度和溴濃度對電池放電性能的影響
本文改變電解液中氫溴酸和溴的濃度,研究了氫溴酸和溴傳質對氫溴電池性能的影響。將電解液中氫溴酸濃度由0.5 mol·L-1提高至1 mol·L-1,進行單電池測試,并計算電池的能量效率、庫倫效率和電壓效率;向氫溴酸濃度1 mol·L-1的電解液中加入溴素(單質溴,分析純,山東祥瑞化工有限公司),將電解液中溴素濃度提高至0.2和0.4 mol·L-1,進行200 mA·cm-2電流密度電池充放電測試。
2.3 氫溴電池的氫氣出口壓力對電池性能的影響
對于氫溴電池,充電過程電池負極產生氫氣,而放電過程,電池負極消耗氫氣,所以在一個充放電循環中,充電和放電過程對氫氣壓力的要求不同。氫溴電池測試平臺儀表盤所顯示的氫出壓力,實際由氫入口壓力手動調壓閥控制,即通過調節電池的氫氣入口壓力來改變電池的氫氣出口壓力。電解液中氫溴酸和溴素濃度分別是2和0.4 mol·L-1;在充放電循環過程中改變氫出壓力值。表2是氫溴電池在充放電循環過程中的氫出壓力。
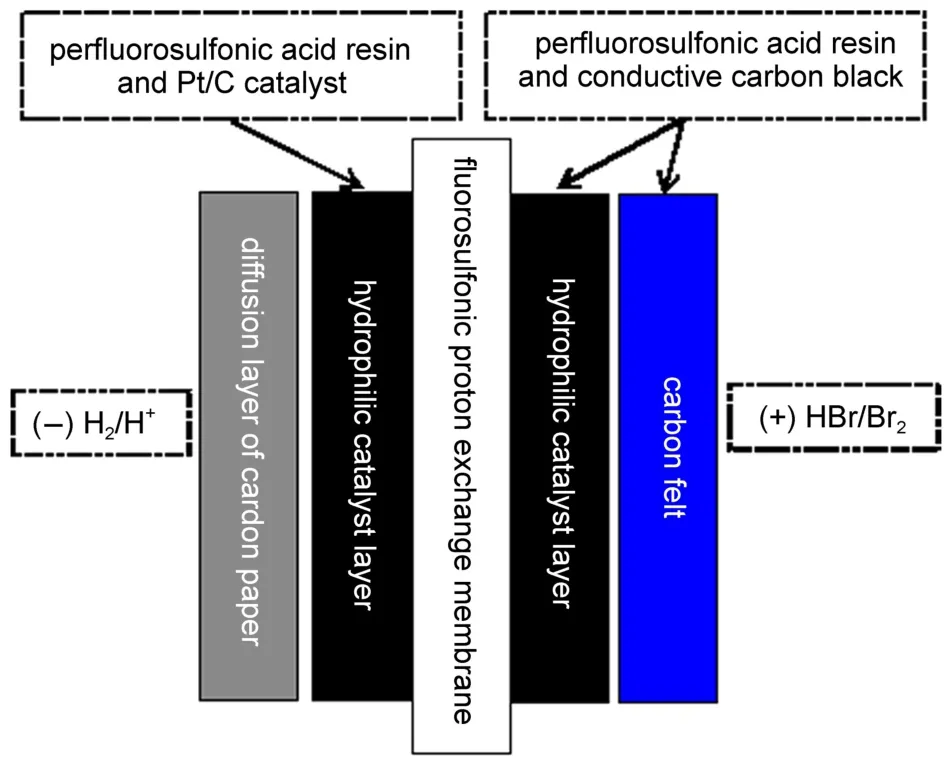
圖2 氫溴電池的第一種電池結構Fig.2 The first structure of the hydrogen bromine battery
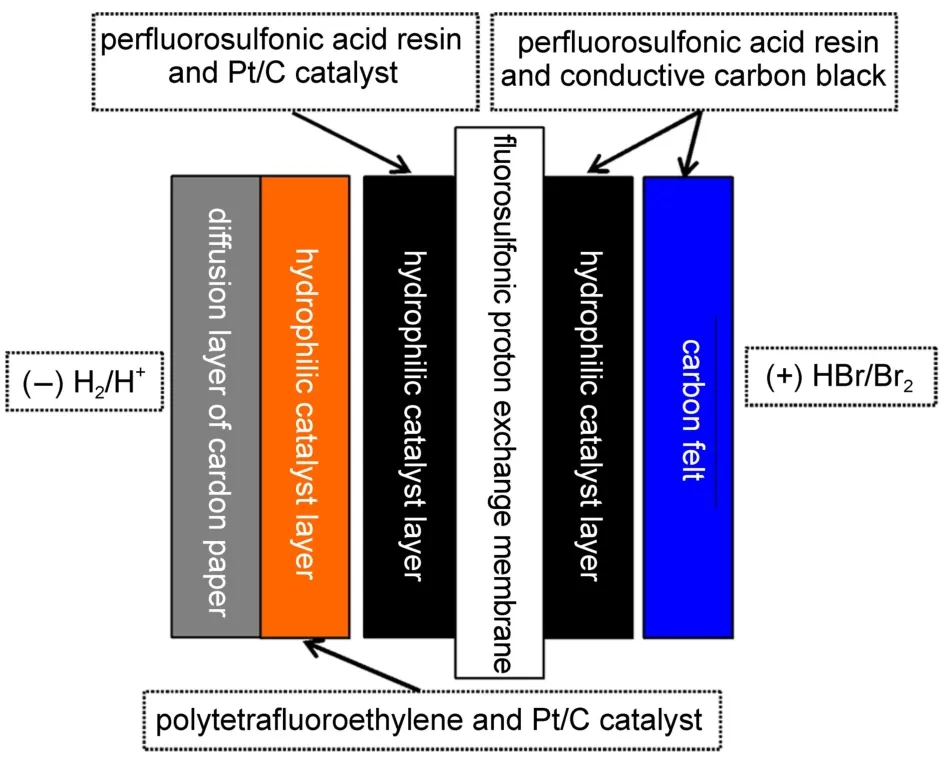
圖3 氫溴電池的第二種電池結構Fig.3 Thesecondstructureofthehydrogenbrominebattery

表1 氫溴單電池測試實驗條件Table 1 Experimental conditions of the hydrogen bromine single cells
2.4 質子交換膜厚度對氫溴電池能量效率的影響
質子交換膜厚度影響電池的膜電阻,尤其在電流密度較大的情況下,膜電壓降很顯著,通過降低電池的膜厚度,可減小電池的內阻電壓降,相應提高氫溴電池的能量效率和電壓效率。本文進一步在兩方面對氫溴電池測試系統進行了改進:一,將電解液組成由2 mol·L-1氫溴酸和0.4 mol·L-1溴大幅提高至3 mol·L-1氫溴酸和5 mol·L-1溴;二,氫溴電池所使用的Nafion膜厚度,由50.0 μm降至20.0和15.0 μm。
2.5 電池能量效率(ηe)、庫倫效率(ηc)、電壓效率(ηv)的計算
(1)能量效率
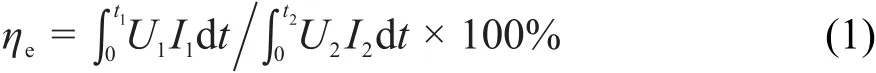
式中:ηe是能量效率;U1、U2分別是放電、充電電壓(V);I1、I2分別是放電、充電電流(A);t1、t2分別是放電、充電時間(s)。
由于恒流放電和恒流充電的電流I1=I2=200 mA·cm-2,所以能量效率為
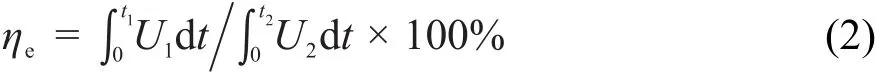
(2)庫倫效率
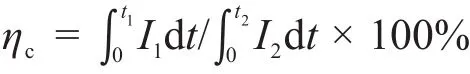
由于恒流放電和恒流充電的電流I1=I2=200 mA·cm-2,所以庫倫效率為

(3)電壓效率


表2 氫溴單電池的氫出壓力Table 2 Hydrogen outlet pressure of the hydrogen bromine single cells
3 結果與討論
3.1 氫溴電池結構對電池性能的影響
圖4(a)和(b)分別是兩種電池結構的氫溴電池極化曲線和功率曲線。如圖4(a)所示,第二種電池的充電電壓較第一種電池明顯降低,放電電壓較第一種電池略有升高,在電流密度為200 mA·cm-2時,第二種電池的充電電壓較第一種電池降低117 mV,放電電壓高于第一種電池3 mV;由圖4(b)功率密度曲線,也反映出這種電池結構的改變,對充電過程影響最大,電流密度越大,充電電壓之間的區別越顯著,而對放電過程影響較小。
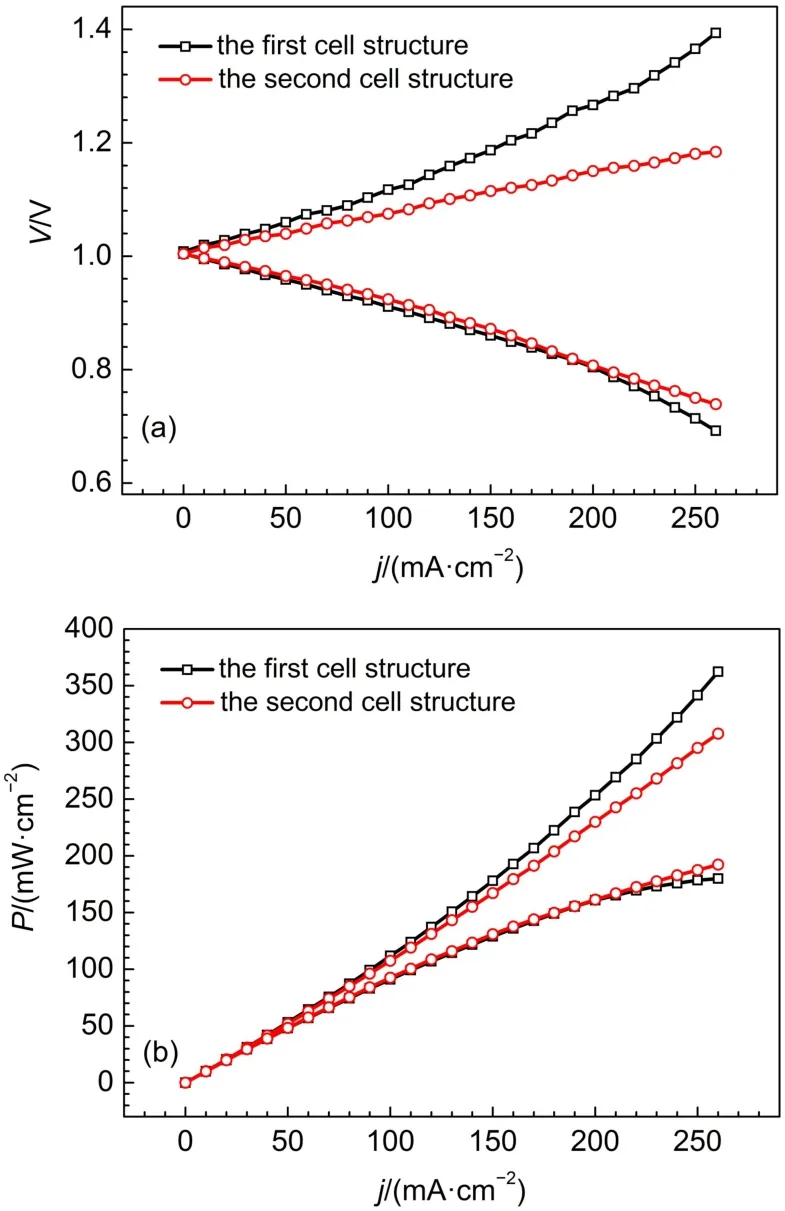
圖4 兩種電池結構的氫溴電池極化曲線(a)和功率曲線(b)Fig.4 Polarization curves(a)and power curves(b)of the hydrogen bromine battery with different cell structures
充電過程,氫離子由正極向負極傳遞,隨氫離子共同傳遞的還有氫溴酸和溴,這導致質子交換膜負極的氫離子還原有效催化面積減小,并且透過的酸液還會對鉑催化劑造成腐蝕,使催化劑的電化學比表面積下降。而第二種電池的碳紙擴散層由于具有憎水性,加上氫氣的吹掃作用,利于膜透酸液的排出,有效降低了酸液滲透所造成的負面作用,并且利于氫氣的脫附,所以第二種電池的充電電壓較第一種電池大幅降低了一百多毫伏。放電過程中,氫離子經膜由負極向正極傳遞,膜透酸量甚微,并且由于氫氣的吹掃作用,膜透酸易于排出,第二種電池的放電電壓略高于第一種電池,主要原因是第二種電池結構具有憎水性,利于氫氣的傳質29。
由圖4(a)極化曲線,第一種電池結構的電池,在電流密度200 mA·cm-2時,充電電壓1.267 V,放電電壓0.804 V,由于充電電壓大于1.2 V,所以該電池不適合在200 mA·cm-2充電運行。而第二種電池結構的電池,在電流密度200 mA·cm-2時,充電電壓1.150 V,放電電壓0.807 V,可以在200 mA·cm-2電流密度下運行。圖5是在200 mA·cm-2電流密度下,由第二種電池組裝的單電池的恒流充放電曲線,由公式(2-4),計算出該電池前三次恒流充放電的能量效率和電壓效率分別為49.2%、44.2%、42.8%。由于充放電電流和充放電時間均相同,所以電池的庫倫效率為100%,這正是由于溴電極電化學反應高度可逆,電化學反應速率快的原因。
3.2 電解液中氫溴酸濃度和溴濃度對電池放電性能的影響
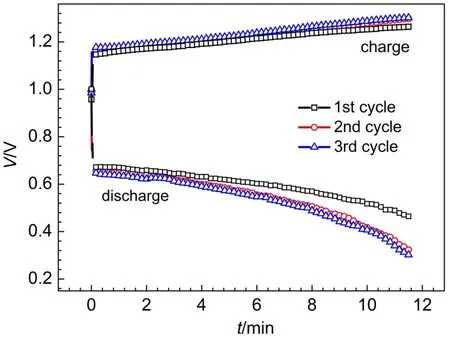
圖5 在電流密度為200 mA·cm-2時第二種電池結構的氫溴電池恒流充放電曲線Fig.5 Charge and discharge curves of the hydrogen bromine battery with the second kind of cell structure at 200 mA·cm-2current density
采用第二種電池結構,將電解液中氫溴酸濃度由0.5 mol·L-1提高至1 mol·L-1,組裝單電池進行測試。如圖6(a)所示,在電流密度200 mA·cm-2,電解液為0.5 mol·L-1氫溴酸的電池,充電電壓為1.150 V,當電解液濃度提高至1 mol·L-1,充電電壓降低了80 mV。這是因為溴電極電化學反應速度快,電極界面附近氫溴酸濃度會迅速降低,容易出現濃差極化,而提高電解液中氫溴酸濃度,加快了氫溴酸傳質速率,降低了溴電極的濃差極化。由放電極化曲線,電流密度小于170 mA·cm-2,電解液濃度為0.5 mol·L-1HBr的氫溴電池放電電壓較高。在大電流密度放電時,電解液為1 mol·L-1HBr的電池性能較好,在電流密度為200 mA·cm-2時,放電電壓為0.825 V,較電解液濃度為0.5 mol·L-1HBr的電池,放電電壓提高了18 mV。圖6(b)是電流密度200 mA·cm-2,兩種電解液濃度的氫溴電池恒流充放電曲線,經計算,1 mol·L-1HBr電池的能量效率和電壓效率為77.1%,較0.5 mol·L-1HBr氫溴酸電池的能量效率和電壓效率提高27.9%。
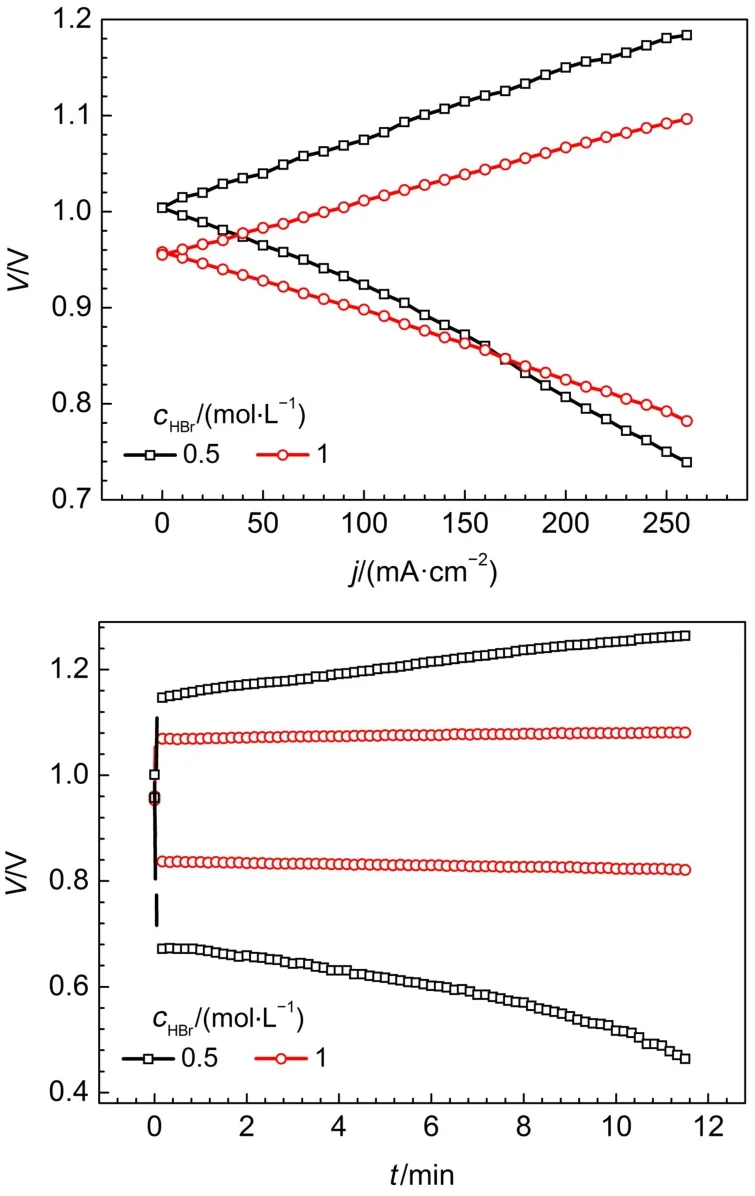
圖6 在電流密度為200 mA·cm-2時氫溴電池極化曲線(a)和恒流充放電曲線(b)Fig.6 Polarization curves(a)and charge and discharge curves(b)of the hydrogen bromine battery at 200 mA·cm-2current density
放電過程的電壓高低不僅取決于電解液中的溴濃度,也與氫溴酸濃度和溴在電解液中的傳質快慢有關。同樣在200 mA·cm-2電流密度下充電相同時間,1 mol·L-1氫溴酸和0.5 mol·L-1氫溴酸所生成的溴物質的量是相同的,在平衡狀態下,由能斯特方程,1 mol·L-1氫溴酸電解液的電極電勢將低于0.5 mol·L-1氫溴酸電解液的電極電勢;在開路狀態下,以1 mol·L-1氫溴酸作為電解液的電池電壓低于0.5 mol·L-1氫溴酸為電解液的電池電壓,如圖6(a)所示。由于氫溴酸是溴的良好溶劑,溴在1 mol·L-1氫溴酸中的溶解性高于0.5 mol·L-1的氫溴酸,所以溴在1 mol·L-1氫溴酸電解液中向電極的傳質較快。在低電流密度區,電化學反應速率較慢,不會出現濃差極化;而在高電流密度區,電極界面溴反應速率加快,濃差極化影響嚴重30,所以在圖6(a)極化曲線中,0.5 mol·L-1氫溴酸電池的電壓在大電流密度區較低。圖6(b)中,溴在1 mol·L-1氫溴酸電解液中溶解度較高,傳質較快,濃差極化較小,所以電池放電電壓較高;隨放電時間延長,也發現0.5 mol·L-1氫溴酸電池的電壓下降趨勢變快,出現了明顯的濃差極化。Huskinson和Aziz31建立了氫溴電池電極極化模型,認為溴電極的電化學極化和傳質極化是影響電池性能的關鍵因素,在高電流密度下,溴電極的傳質極化影響更大,電解液濃度升高,傳質極化緩解。
電解液中氫溴酸濃度為1 mol·L-1,向電解液中添加單質溴,使電解液中溴濃度提高至0.2和0.4 mol·L-1,在200 mA·cm-2電流密度下進行恒流充放電測試,如圖7所示。電解液中氫溴酸濃度不變,加入溴素,對電池的充電性能影響很小。但電池的放電電壓變化明顯,添加0.4 mol·L-1單質溴的電池,放電性能明顯高于單質溴濃度為0.2 mol·L-1和無溴的單電池。計算電池的能量效率,單質溴濃度由零提高至0.2、0.4 mol·L-1,電池的能量效率分別是77.1%、78.2%和79.0%。
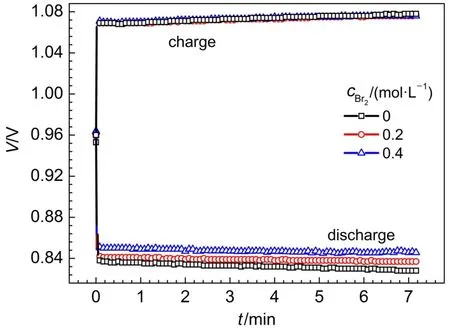
圖7 在電流密度為200 mA·cm-2時改變電解液中單質溴濃度的氫溴電池恒流充放電曲線Fig.7 Charge and discharge curves of the hydrogen bromine battery at 200 mA·cm-2current density with different bromine concentration in the electrolyte
電解液中氫溴酸濃度是影響電池充電電壓高低的主要因素,所以向電解液中添加單質溴,并沒有顯著影響充電過程。而對于放電過程,向電解液中添加單質溴,溴電極電化學還原反應速度加快、電化學極化減小,同時溴傳質加快,減小了濃差極化,所以電池的放電電壓提高,電池的能量效率和電壓效率均明顯提高32。
3.3 氫溴電池的氫氣出口壓力對電池性能的影響
電解液中氫溴酸濃度為2 mol·L-1,溴濃度為0.4 mol·L-1,在電流密度為200 mA·cm-2進行電池恒流充放電測試,在充放電過程的不同步驟中,采用不同的氫出壓力,實驗結果如圖8所示。
由圖8數據計算出每個循環在不同氫出壓力下電池的能量效率,如圖9所示。可見在充電過程中,隨氫出壓力的減小,電池能量效率顯著提高,氫出壓力為零時,電池能量效率達到80.0%。這是因為充電過程中負極產生氫氣,降低電池的氫出壓力,有利于負極氫氣的產生、逸出,降低催化劑表面氫氣的吸附,相應增大了催化劑的有效催化面積。但是充電過程中,降低氫出壓力,這將增大電池正負極之間的壓力差,使質子交換膜由正極向負極的透酸現象更嚴重。膜透酸使負極Pt/C催化劑性能衰減,不利于電池的長期穩定運行,所以充電過程中,負極需要維持一定的氫出壓力,以解決膜透酸的問題。
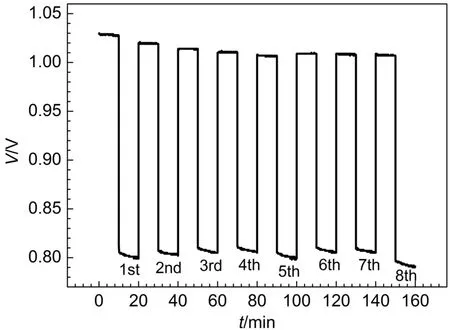
圖8 在電流密度為200 mA·cm-2時不同氫出壓力下氫溴電池恒流充放電測試Fig.8 Charge and discharge experiment for the hydrogen bromine battery under different hydrogen pressures at 200 mA·cm-2current density
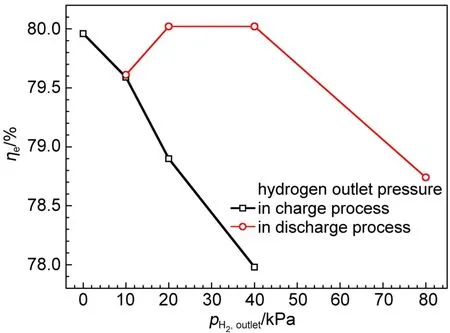
圖9 氫出壓力對氫溴電池能量效率的影響Fig.9 Effect of hydrogen pressure on the energy efficiency(ηe)of the hydrogen bromine battery
在放電過程中,氫出壓力過大或過小,電池的能量效率均較低。氫出壓力小,將影響氫氣傳質,使濃差極化增大,放電電壓降低,相應電池效率會降低。而氫氣壓力過大,由于氫氣的電化學氧化反應速度有限,將增大氫氣在催化劑表面的吸附,影響催化層的質子傳導。放電過程,理想的氫氣壓力是可維持氫氣在催化劑表面的單層吸附,所以放電過程中,氫出壓力控制在20.0-40.0 kPa較合適,此范圍的能量效率和電壓效率為80.0%。經反復實驗,確定在氫溴電池充電和放電過程中,電池的氫出壓力均控制在40.0 kPa;并且氫溴電池測試平臺的氫出口尾排閥全開,可以滿足氫溴電池在充電和放電過程中對氫氣壓力控制的不同需求。始終維持氫溴電池氫側有一定的壓力,這樣可以延遲或避免質子交換膜透酸現象的發生,并且使電池具有較高的效率。
3.4 質子交換膜厚度對氫溴電池能量效率的影響
電解質溶液中氫溴酸和溴濃度分別是3和5 mol·L-1,分別使用50.0、20.0和15.0 μm的質子交換膜組裝氫溴單電池,進行充放電性能測試。圖10(a)和(b)分別是三種膜厚度的氫溴單電池極化曲線和功率曲線,由圖10(a),膜厚度由50.0 μm降至20.0 μm,充電電壓明顯下降,這正是由于膜減薄后膜電阻降低的原因。膜厚度降低至15.0 μm,發現電池的充電電壓較20.0 μm的Nafion膜又明顯增大,這是由于膜薄,雖然膜電阻會降低,但是膜由正極向負極透酸現象更嚴重,造成Pt/C催化劑性能衰減,電化學極化增大,充電電壓反而升高。
比較放電過程,在小電流密度放電時,降低膜厚度,雖可降低膜電阻極化,但此時,膜透酸所造成的負極Pt/C催化劑性能衰減是影響電池性能的主要因素,厚膜對于阻止膜透酸更具優勢,所以具有更高的放電電壓。只有在大電流密度放電時,如超過200 mA·cm-2,膜電阻極化成為影響電池放電性能的主要因素,所以,此時薄的質子交換膜具有更高的放電電壓和功率密度,如圖10 (b)所示。
所以氫溴電池在超過200 mA·cm-2電流密度運行時,降低膜厚度有效降低膜電阻極化,對放電過程更有利。但充電過程,降低膜厚度,隨氫離子的傳遞,膜透酸現象更嚴重,所以膜不能太薄,厚度20.0 μm的質子交換膜更有利于充電過程。Tucker等33認為膜的性質對于氫溴電池的性能和效率具有重要影響,電池的運行應權衡膜的導電性和溴與水分子的穿流兩因素,在高電流密度區,導電性制約電池效率的提升;而在低電流密度區,溴和水分子的穿流限制了電池效率的提高。
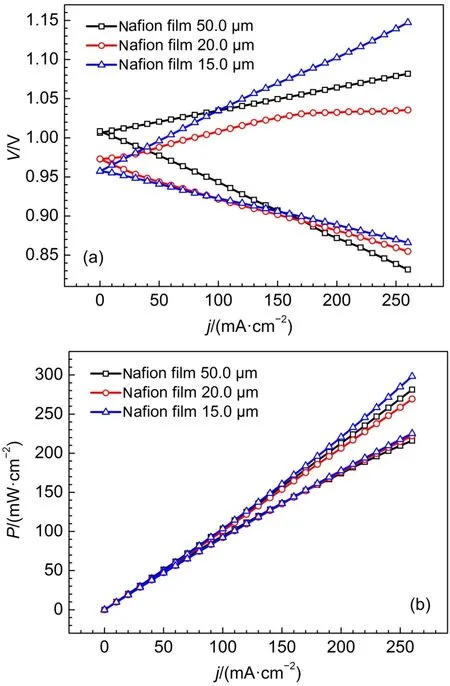
圖10 不同膜厚度的氫溴單電池測試極化曲線(a)和功率曲線(b)Fig.10 Polarization curves(a)and power curves(b)of the hydrogen bromine battery with different membrane thickness
圖11(a,b,c)分別是在電流密度為200 mA·cm-2時,分別使用50.0、20.0和15.0 μm質子交換膜的氫溴電池,前五次恒流充放電曲線,如圖11(a)和圖11(b)所示。隨循環次數增加,50.0 μm Nafion膜和20.0 μm Nafion膜的氫溴電池充電電壓下降,這可能是由于隨循環次數的增加,溴電極表面潤濕程度增大,尤其是碳氈的多孔網絡結構與氫溴酸電解液充分接觸,催化反應活性面積增大,電化學反應速度加快,電化學極化和濃差極化均減小所致。而如圖11(c)所示,隨循環次數增加,充電電壓升高,這可能是由于膜太薄,正極向負極透酸現象嚴重,負極Pt/C催化劑的催化活性下降,負極電化學極化增大所致。在放電過程中,三種膜厚度的電池,均表現出隨循環次數增加,放電電壓下降的趨勢,這是由于隨循環次數增加,溴電極表面的不可逆吸附,會使電極的有效催化活性面積下降,電化學極化增大;并且膜透酸也會造成負極Pt/C催化劑腐蝕,使負極電化學比表面積下降,電化學極化增大。

圖11 不同質子交換膜厚度的氫溴電池恒流充放電曲線Fig.11 Charge and discharge curves of the hydrogen bromine battery with different thickness proton exchange membrane
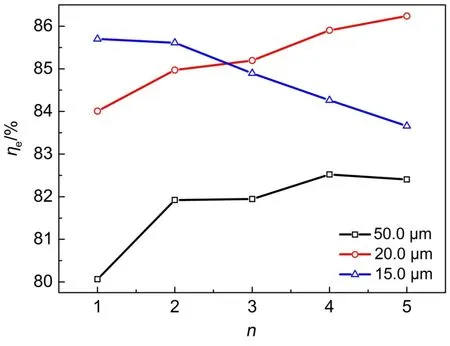
圖12 不同厚度膜的氫溴電池能量效率曲線Fig.12 Energy efficiency of the hydrogen bromine battery with different thickness of the membrane
由恒流充放電曲線,計算出三種膜厚度的氫溴電池前五次循環的能量效率,如圖12所示。20.0 μm Nafion膜氫溴電池的前五次循環平均能量效率為85.3%,高于15.0 μm Nafion膜的氫溴電池,50.0 μm Nafion膜的氫溴電池前五次循環平均能量效率最低,僅為81.8%。Livshits等32以納米多孔質子傳導膜為隔膜,電解液1 mol·L-1HBr和0.9 mol·L-1Br2,電極面積5 cm2,溴電極負載1 mg 30%Pt/C,電池的能量效率接近90%。本實驗的膜電極面積為186 cm2,溴電極碳氈負載的是導電炭黑BP 2000,對溴的催化能力遠低于Pt/C催化劑,所以電池的能量效率僅達到約85%。
對于50.0和20.0 μm Nafion膜的氫溴電池,發現隨循環次數增加,前五次循環能量效率有增大的趨勢,這是因為雖然循環次數增加,溴電極的不可逆吸附也會增大,但是溴電極的潤濕性也在逐漸改善。如圖11(a)和(b),恒流放電前五次循環,雖然電池放電性能逐漸降低,但電池充電性能卻在逐漸提高,表現在充電電壓逐漸降低,所以50.0和20.0 μm Nafion膜的氫溴電池,前五次循環,電池的能量效率是提高的。對于15.0 μm Nafion膜氫溴電池,雖然循環次數增大,溴電極碳氈的潤濕性也在逐漸改善,但是由于膜太薄,正極向負極的膜透酸現象造成負極Pt/C催化劑電化學活性面積下降明顯,這是電池能量效率降低的主要原因。
4 結論
(1)在氫溴電池負極碳紙擴散層上建立Pt/C憎水催化層,有助于膜透酸的氫氣吹掃排出,提高Pt/C催化劑的電化學活性比表面積;在正極碳氈上浸漬導電炭黑BP2000,提高溴電極的電化學活性比表面積,這是氫溴電池可在200 mA·cm-2電流密度下,持續穩定運行的關鍵。
(2)提高電解液中氫溴酸濃度,可減小充電過程的正極濃差極化,氫溴電池充電性能提高;同時,提高電解液中氫溴酸濃度,有利于提高溴的溶解性,降低放電過程中溴電極的濃差極化,氫溴電池放電性能提高;向氫溴酸中添加單質溴,也有利于降低放電過程正極的濃差極化。
(3)降低充電過程中氫出壓力,有利于負極氫氣的產生、逸出,提高電池充電性能,但正負極壓差增大,膜透酸問題會嚴重;放電過程,合適的氫出壓力是維持氫氣在碳紙憎水催化層的單層吸附,有利于質子的傳導;循環過程,充放電均維持氫出壓力在40.0 kPa,可避免壓力頻繁波動對膜電極造成的損壞,同時阻止膜透酸現象的發生。
(4)降低質子交換膜厚度,將降低氫溴電池的內阻電壓降,但是當膜厚度降低到一定程度,充電過程中膜透酸問題會很嚴重,使負極Pt/C催化劑電化學活性比表面積下降,電池充電性能降低;膜厚度對放電過程的影響與電流密度有關,低電流密度區,膜透酸使負極電化學活性比表面下降居主導地位,這是薄膜放電性能較低的主要原因;而在高電流密度區,膜內阻電壓降居主導地位,薄膜內阻低,電壓降較小,薄膜放電性能較高。
(5)采用20.0 μm質子交換膜,電解液中氫溴酸濃度和溴濃度分別為3和5 mol·L-1,氫出壓力40.0 kPa,在200 mA·cm-2電流密度恒流充放電,前五次循環,電池的能量效率和電壓效率達到85.3%,庫倫效率達到100%。
(1) Panwar,N.L.;Kaushik,S.C.;Kothari,S.Renewable Sustainable Energy Rev.2011,15(3),1513.doi:10.1016/j. rser.2010.11.037
(2) Zeng,Y.K.;Zhou,X.L.;An,L.;Wei,L.;Zhao,T.S.J.Power Sources 2016,324,738.doi:10.1016/j.jpowsour.2016.05.138
(3) Zeng,Y.K.;Zhao,T.S.;An,L.;Zhou,X.L.;Wei,L.J.Power Sources 2015,300,438.doi:10.1016/j.jpowsour.2015.09.100
(4)Wei,L.;Zhao,T.S.;Zhao,G.;An,L.;Zeng,L.Appl.Energy 2016,176,74.doi:10.1016/j.apenergy.2016.05.048
(5) Roznyatovskaya,N.;Noack,J.;Fühl,M.;Pinkwart,K.;Tübke, J.Electrochim.Acta 2016,211,926.doi:10.1016/j. electacta.2016.06.073
(6) Zhang,L.Q.;Lai,Q.Z.;Zhang,J.L.;Zhang,H.M. ChemSusChem 2012,5(5),867.doi:10.1002/cssc.201100530
(7) Yang,H.S.;Park,J.H.;Ra,H.W.;Jin,C.S.;Yang,J.H.J. Power Sources 2016,325,446.doi:10.1016/j. jpowsour.2016.06.038
(8) Zhao,P.;Zhang,H.M.;Zhou,H.T.;Yi,B.L.Electrochim.Acta 2005,51(6),1091.doi:10.1016/j.electacta.2005.06.008
(9) Zhou,H.T.;Zhang,H.M.;Zhao,P.;Yi,B.L.Electrochim.Acta 2006,51(28),6304.doi:10.1016/j.electacta.2006.03.106
(10) Wei,X.L.;Li,L.Y.;Luo,Q.T.;Nie,Z.M.;Wang,W.;Li,B.; Xia,G.G.;Miller,E.;Chambers,J.;Yang,Z.G.J.Power Sources 2012,218,39.doi:10.1016/j.jpowsour.2012.06.073
(11) Li,B.;Li,L.Y.;Wang,W.;Nie,Z.M.;Chen,B.W.;Wei,X.L.; Luo,Q.T.;Yang,Z.G.;Sprenkle,V.J.Power Sources 2013, 229,1.doi:10.1016/j.jpowsour.2012.11.119
(12) http://www.tau.ac.il/institutes/ifcbc/pdf/_EnStorage% 20presentation%20%20Public%206-07.pdf(accessed June, 2007).
(13) Lin,G.;Chong,P.Y.;Yarlagadda,V.;Nguyen,T.V.;Wycisk,R. J.;Pintauro,P.N.;Bates,M.;Mukerjee,S.;Tucker,M.C.; Weber,A.Z.J.Electrochem.Soc.2016,163(1),A5049. doi:10.1149/2.0071601jes
(14) Cho,K.T.;Tucker,M.C.;Ding,M.;Ridgway,P.;Battaglia,V. S.;Srinivasan,V.;Weber,A.Z.ChemPlusChem 2015,80(2), 402.doi:10.1002/cplu.201402043
(15) Wang,C.H.;Li,X.F.;Xi,X.L.;Zhou,W.;Lai,Q.Z.;Zhang, H.M.Nano Energy 2016,21,217.doi:10.1016/j. nanoen.2016.01.015
(16) Zhang,L.Q.;Zhang,H.M.;Lai,Q.Z.;Li,X.F.;Cheng,Y.H. J.Power Sources 2013,227,41.doi:10.1016/j. jpowsour.2012.11.033
(17) H?hne,K.;Starbeck,G.Hydrogen/Bromine Cell.U.S.Pat. Appl.4520081,1985.
(18) Braff,W.A.;Bazant,M.Z.;Buie,C.R.Nat.Commun.2013,4, 2346.doi:10.1038/ncomms3346
(19) Xu,W.;Scott,K.Int.J.Hydrog.Energy 2010,35(21),12029. doi:10.1016/j.ijhydene.2010.08.055
(20) Medina,P.;Santarelli,M.Int.J.Hydrog.Energy 2010,35(11), 5173.doi:10.1016/j.ijhydene.2010.02.130
(21) Ehteshami,S.M.M.;Vignesh,S.;Rasheed,R.K.A.;Chan,S. H.Appl.Energy 2016,170,388.doi:10.1016/j. apenergy.2016.03.001
(22) Mikkola,M.S.;Rockward,T.;Uribe,F.A.;Pivovar,B.S.Fuel Cells 2007,7(2),153.doi:10.1002/fuce.200600206
(23) Hongsirikarn,K.;Goodwin,J.G.,Jr.;Greenway,S.;Creager,S. J.Power Sources 2010,195(21),7213.doi:10.1016/j. jpowsour.2010.05.005
(24) Jie,X.;Shao,Z.G.;Yi,B.L.Electrochem.Commun.2010,12 (5),700.doi:10.1016/j.elecom.2010.03.010
(25) Park,J.W.;Wycisk,R.;Pintauro,P.N.J.Membr.Sci.2015, 490,103.doi:10.1016/j.memsci.2015.04.044
(26) Peled,E.;Blum,A.;Goor,M.Encyclopedia of Electrochemical Power Sources,1st ed.;Elservier:Amsterdam,2009;pp 182-191.doi:10.1016/B978-044452745-5.00860-1
(27) Costa,R.F.D.;Ferreira,J.Z.;Deslouis,C.J.Membr.Sci.2003, 215(1-2),115.doi:10.1016/S0376-7388(02)00607-5
(28) Saito,M.;Ikesaka,S.;Kuwano,J.;Qiao,J.;Tsuzuki,S.; Hayamizu,K.;Okada,T.Solid State Ionics 2007,178(7-10), 539.doi:10.1016/j.ssi.2007.01.001
(29) T?tzke,C.;Gaiselmann,G.;Osenberg,M.;Arlt,T.;Mark?tter, H.;Hilger,A.;Kupsch,A.;Müller,B.R.;Schmidt,V.;Lehnert, W.;Manke,I.J.Power Sources 2016,324,625.doi:10.1016/j. jpowsour.2016.05.118
(30) Cho,K.T.;Ridgway,P.;Weber,A.Z.;Haussener,S.;Battaglia, V.;Srinivasan,V.J.Electrochem.Soc.2012,159(11),A1806. doi:10.1149/2.018211jes
(31) Huskinson,B.;Aziz,M.J.Energy Sci.Technol.2013,5(1),1. doi:10.3968/j.est.1923847920130501.854
(32) Livshits,V.;Ulus,A.;Peled,E.Electrochem.Commun.2006,8 (8),1358.doi:10.1016/j.elecom.2006.06.021
(33) Tucker,M.C.;Cho,K.T.;Spingler,F.B.;Weber,A.Z.;Lin,G. Y.J.Power Sources 2015,284,212.doi:10.1016/j. jpowsour.2015.03.010
Hydrogen Bromine Battery Structure Optimization and the Operation Condition Effects on Battery Performance
SHI Ji-Cheng1XU Hong-Feng1,*LU Lu1GAO Jun2
(1Liaoning Province Key Laboratory of New Energy Battery,College of Environmental and Chemical Engineering,Dalian Jiaotong University,Dalian 116028,Liaoning Province,P.R.China;2Heilongjiang Daqing Refining&Chemical Company, Daqing 163411,Heilongjiang Province,P.R.China)
This paper studies how particular factors affect hydrogen bromine batteries,including the cell structure,the hydrobromic acid and bromine concentrations,the hydrogen pressure and the proton exchange membrane thickness.After the Pt/C hydrophobic catalyst layer was loaded onto the carbon paper,the hydrogen bromine battery worked at 200 mA·cm-2current density and the battery Coulombic efficiency was 100%.The bromine electrode electrochemical reaction was controlled by the concentration polarization.The battery performance improved when the hydrobromic acid concentration increased.The bromine solubility also increased at the higher hydrobromic acid concentration and the battery discharge performance improved.When the hydrobromic acid concentration was increased from 0.5 mol·L-1to 1 mol·L-1,the energy efficiency and the voltage efficiency increased by 27.9%at the current density of 200 mA·cm-2.In the charge process,asthe hydrogen pressure was reduced,the battery charging performance improved,but severe membrane acid permeability was observed.In the discharge process,the optimal hydrogen pressure was able to maintain the monolayer hydrogen adsorption on the hydrophobic catalyst layer on the carbon paper.The energy efficiency was 80.2%at 40.0 kPa hydrogen pressure in the charge and discharge processes.In the charge process,the membrane thickness was closely related to the membrane resistance polarization and the membrane acid permeability.After the membrane thickness was reduced from 50.0 to 15.0 μm,acid permeability through the membrane was more severe.This reduced the electrochemical active surface area and a reduction in the battery performance was observed.In the discharge process,the membrane acid permeability was the leading factor at the lower current density;the battery with the 50.0 μm Nafion membrane had a higher discharge performance.As the current density was>200 mA·cm-2,the membrane polarization resistance was the dominated factor;the battery with the 15.0 μm Nafion membrane had a higher discharge performance.With the 20.0 μm proton exchange membrane,the energy efficiency and voltage efficiency of the hydrogen bromine battery were 85.3%and the Coulombic efficiency was 100%at the current density of 200 mA·cm-2with five cycles.
Hydrogen bromine battery;Membrane electrode;Proton exchange membrane;Membrane acid permeability; Battery efficiency
O646
10.3866/PKU.WHXB201609195
Received:July 20,2016;Revised:September 19,2016;Published online:September 19,2016.
*Corresponding author.Email:hfxu@fuelcell.com.cn;Tel:+86-411-84106713.
The project was supported by the National Natural Science Foundation of China(21376034,21406024)and National High-Tech Research and Development Program,China(863)(2012AA052002).
國家自然科學基金(21376034,21406024)和國家高技術研究發展計劃(863)(2012AA052002)資助項目
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
