氣相色譜法同時測定復方氨酚烷胺片中4種成分的含量
2017-03-29 09:56:32董秋香張月寒付萍萍劉志瑋保定市食品藥品檢驗所河北保定071051
中國藥房 2017年6期
董秋香,張月寒,付萍萍,劉志瑋(保定市食品藥品檢驗所,河北保定 071051)
氣相色譜法同時測定復方氨酚烷胺片中4種成分的含量
董秋香*,張月寒,付萍萍,劉志瑋(保定市食品藥品檢驗所,河北保定 071051)
目的:建立同時測定復方氨酚烷胺片中對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏含量的方法。方法:采用氣相色譜法。色譜柱為HP-5石英毛細管柱,程序升溫,檢測器為氫火焰離子化檢測器,檢測器溫度為300℃,載氣為氮氣,流速為1.5 mL/min,分流比為20∶1,進樣量為1 μL。結果:對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏檢測質量濃度線性范圍分別為156.0~4 990.4、125.7~4 023.2、19.14~612.4、2.515~80.48 μg/mL(r均為0.999 9);定量限分別為1.4、0.5、1.1、0.9 ng,檢測限分別為0.4、0.2、0.3、0.3 ng;精密度、穩定性、重復性試驗的RSD<2.0%;加樣回收率分別為99.59%~101.77%(RSD=0.8%,n=9)、99.56%~101.80%(RSD=0.7%,n=9)、98.44%~100.83%(RSD=0.7%,n=9)、100.05%~101.91%(RSD=0.6%,n=9)。結論:該方法簡便快速、準確可靠,適用于復方氨酚烷胺片中對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的同時測定。
復方氨酚烷胺片;氣相色譜法;對乙酰氨基酚;鹽酸金剛烷胺;咖啡因;馬來酸氯苯那敏;含量
復方氨酚烷胺片是常見的抗感冒藥,用于緩解普通感冒及流行性感冒引起的發熱、頭痛、鼻塞、咽痛等癥狀,也可用于流行性感冒的預防和治療。本品為復方制劑,由對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏及人工牛黃組成,收載于《國家藥品標準》[1]和國家食品藥品監督管理局標準YBH06802005[2]。現行標準中的含量測定分別采用滴定法和高效液相色譜法(HPLC);滴定法操作煩瑣,易產生誤差,結果重復性差。該制劑中各成分的處方量、紫外吸收強度差異均較大,采用HPLC法測定其中主成分的含量,多為測定對乙酰氨基酚、咖啡因或馬來酸氯苯那敏中1~3種成分的含量[3-6],若同時測定4種成分的含量需采用雙波長法[7],無法實現真正意義上的同時檢測。因此,本試驗采用氣相色譜法(GC)建立了同時測定該制劑中對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏含量的方法,以期為全面評價藥品質量提供參考。
1 材料
1.1 儀器
7820A型GC儀,包括氫火焰離子化檢測器、G4513A自動進樣器、EC Chrom Elite工作站(美國Agilent公司);BP211D型電子天平(德國Sartorius公司);DK-2000-ⅢLA型電熱恒溫水浴鍋(天津泰斯特儀器有限公司)。
1.2 藥品與試劑
復方氨酚烷胺片(A廠,批號:141028、141023、140414、140931、20130613、141020、20141007;B廠,批號:1410087、1410071、1411092;C廠,批號:1501241、1411071;D廠,批號:20150106、20140911;E廠,批號:140701;F廠,批號:141216;G廠,批號:1141241)規格均為12片/盒;鹽酸金剛烷胺對照品(批號:100426-201002,純度:100%)、對乙酰氨基酚對照品(批號:100018-201409,純度:99.9%)、咖啡因對照品(批號:171215-200406,純度:100%)和馬來酸氯苯那敏對照品(批號:100047-200606,純度:99.7%)均購自中國食品藥品檢定研究院;無水乙醇、三氯甲烷為分析純。
2 方法與結果
2.1 色譜條件
色譜柱:HP-5石英毛細管柱(30 m×0.32 mm,0.25 μm);程序升溫:起始溫度為170℃,保持3 min,再以12℃/min升溫至260℃,保持2 min;檢測器:氫火焰離子化檢測器;檢測器溫度:300℃;載氣:氮氣;流速:1.5 mL/min;分流比:20∶1;進樣量:1 μL。
2.2 溶液的制備
2.2.1 混合對照品溶液 稱取馬來酸氯苯那敏對照品50.30 mg,置于25 mL量瓶中,加無水乙醇溶解并定容,搖勻,作為馬來酸氯苯那敏對照品溶液;稱取對乙酰氨基酚對照品124.76 mg、鹽酸金剛烷胺對照品100.58 mg、咖啡因對照品15.31 mg,置于25 mL量瓶中,加無水乙醇適量使溶解,再精密加入馬來酸氯苯那敏對照品溶液1 mL,加無水乙醇定容,搖勻,作為混合對照品貯備液。取上述混合對照品貯備液適量,置于25 mL量瓶中,加無水乙醇定容,搖勻,作為混合對照品溶液。
2.2.2 供試品溶液 取樣品20片,精密稱定,研細,稱取細粉適量(約相當于對乙酰氨基酚125 mg),置于50 mL量瓶中,加無水乙醇適量,于80℃水浴加熱30 min,放冷至室溫,加無水乙醇定容,搖勻,經0.45 μm微孔濾膜濾過,取續濾液,即得。
2.2.3 陰性對照溶液 按樣品的處方比例和制備工藝制備缺對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的陰性樣品,再按“2.2.2”項下方法制備陰性對照溶液。
2.3 系統適用性試驗
精密量取“2.2”項下混合對照品溶液、供試品溶液和陰性對照溶液各適量,按“2.1”項下色譜條件進樣測定,記錄色譜,詳見圖1。由圖1可知,在該色譜條件下,各成分均能達到基線分離,分離度>10;理論板數以鹽酸金剛烷胺峰計為17 614,保留時間為2.450、5.430、6.870、8.490 min。結果表明,其他成分對測定無干擾。
2.4 線性關系考察
取“2.2.1”項下混合對照品貯備液適量,倍比稀釋,制備乙酰氨基酚質量濃度分別為4 990.4、2 495.2、1 248.6、623.8、311.9、156.0 μg/mL,鹽酸金剛烷胺質量濃度分別為4 023.2、2 011.6、1 005.8、502.9、251.4、125.7 μg/mL,咖啡因質量濃度分別為612.4、306.2、153.1、76.55、38.28、19.14 μg/mL,馬來酸氯苯那敏質量濃度分別為80.48、40.24、20.12、10.06、5.030、2.515 μg/ mL的系列混合對照品溶液。取上述溶液適量,按“2.1”項下色譜條件進樣測定,記錄峰面積。以待測成分質量濃度(x,μg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的回歸方程分別為y=4.404×103x-7.304×104(r=0.999 9)、y=5.734×103x+4.927×104(r=0.999 9)、y=2.597×103x+1.356×102(r=0.999 9)、y=3.383×103x+ 1.789×103(r=0.999 9)。結果表明,對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏檢測質量濃度線性范圍分別為156.0~4 990.4、125.7~4 023.2、19.14~612.4、2.515~80.48 μg/mL。
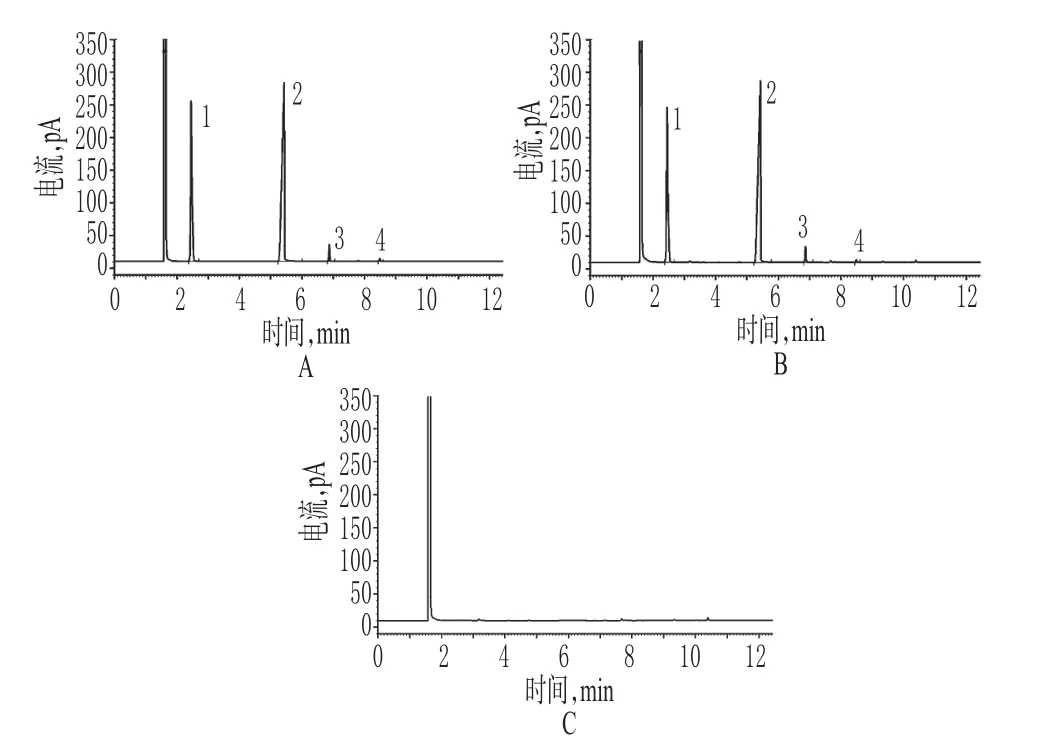
圖1 氣相色譜圖Fig 1 GC Chromatograms
2.5 定量限和檢測限考察
取“2.2.1”項下混合對照品溶液適量,倍比稀釋,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。當信噪比為10∶1時,得定量限(LOQ);當信噪比為3∶1時,得檢測限(LOD)。結果,對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的LOQ分別為1.4、0.5、1.1、0.9 ng;LOD分別為0.4、0.2、0.3、0.3 ng。
2.6 精密度試驗
精密量取“2.2.1”項下混合對照品溶液適量,按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,鹽酸金剛烷胺、對乙酰氨基酚、咖啡因、馬來酸氯苯那敏峰面積的RSD分別為0.2%、0.3%、0.6%、0.4%(n=6),表明儀器精密度良好。
2.7 穩定性試驗
取“2.2.2”項下供試品溶液(批號:141028)適量,分別于室溫下放置0、1、2、4、6、8、10、24 h時按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏峰面積的RSD分別為0.9%、1.2%、1.2%、1.5%(n=8),表明供試品溶液在室溫下放置24 h內穩定性良好。
2.8 重復性試驗
取同一批樣品(批號:141028)適量,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算平均含量。結果,對乙酰氨基酚、鹽酸金剛烷胺、咖啡因和馬來酸氯苯那敏的平均含量分別為99.2%、94.4%、100.7%、98.4%,RSD分別為0.8%、1.0%、0.8%、0.9%(n=6),表明本方法重復性良好。
2.9 加樣回收率試驗
取已知含量的樣品(批號:141028),研細,精密稱取細粉適量(約相當于對乙酰氨基酚125 mg),共9份,各置于50 mL量瓶中,分別精密加入一定質量的對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏對照品,按“2.2.2”項下方法制備低、中、高質量濃度的供試品溶液。取上述溶液適量,按“2.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率,結果見表1。
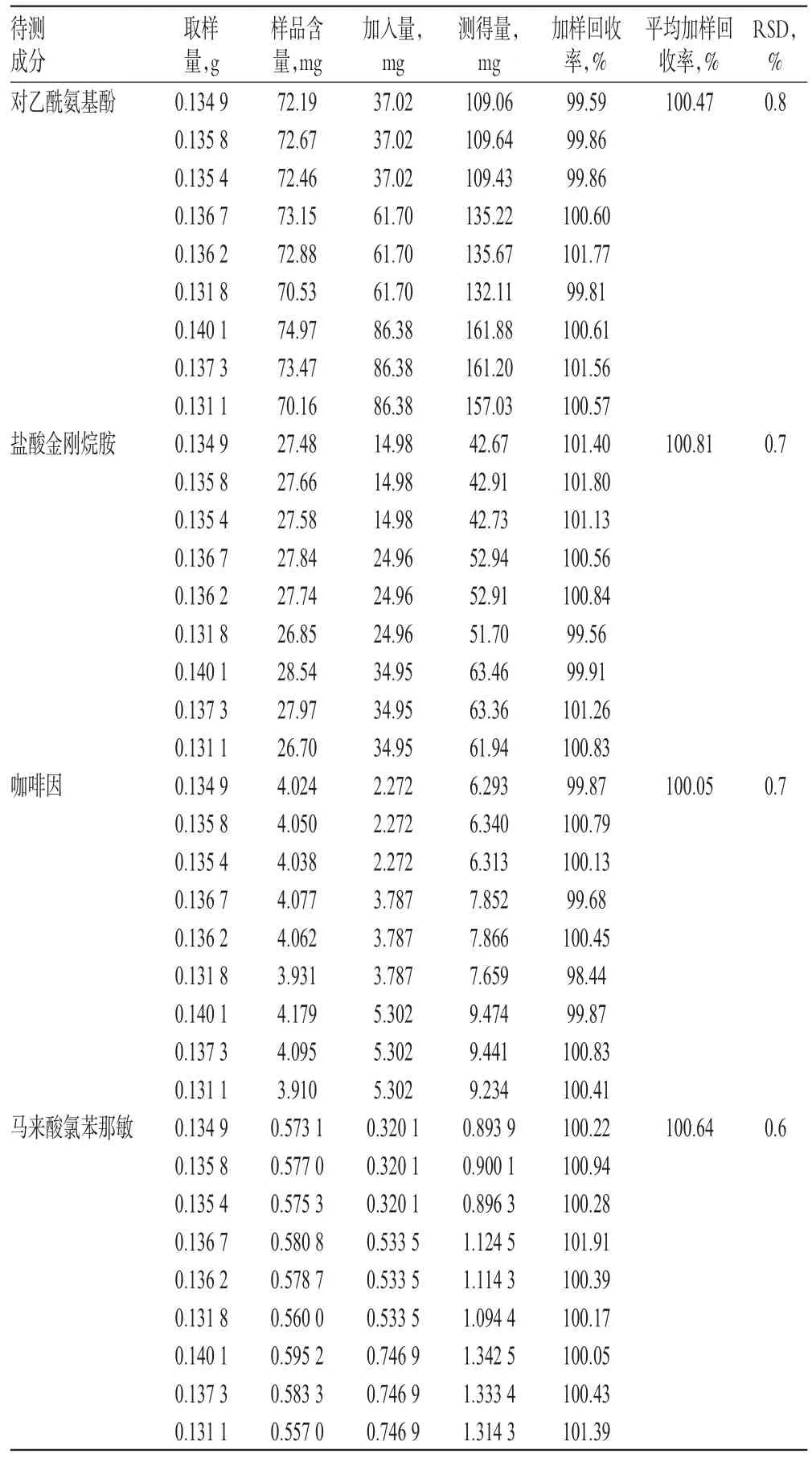
表1 加樣回收率試驗結果(n=9)Tab 1 Results of recovery test(n=9)
2.10 樣品含量測定
取17批樣品各適量,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的含量,結果見表2。
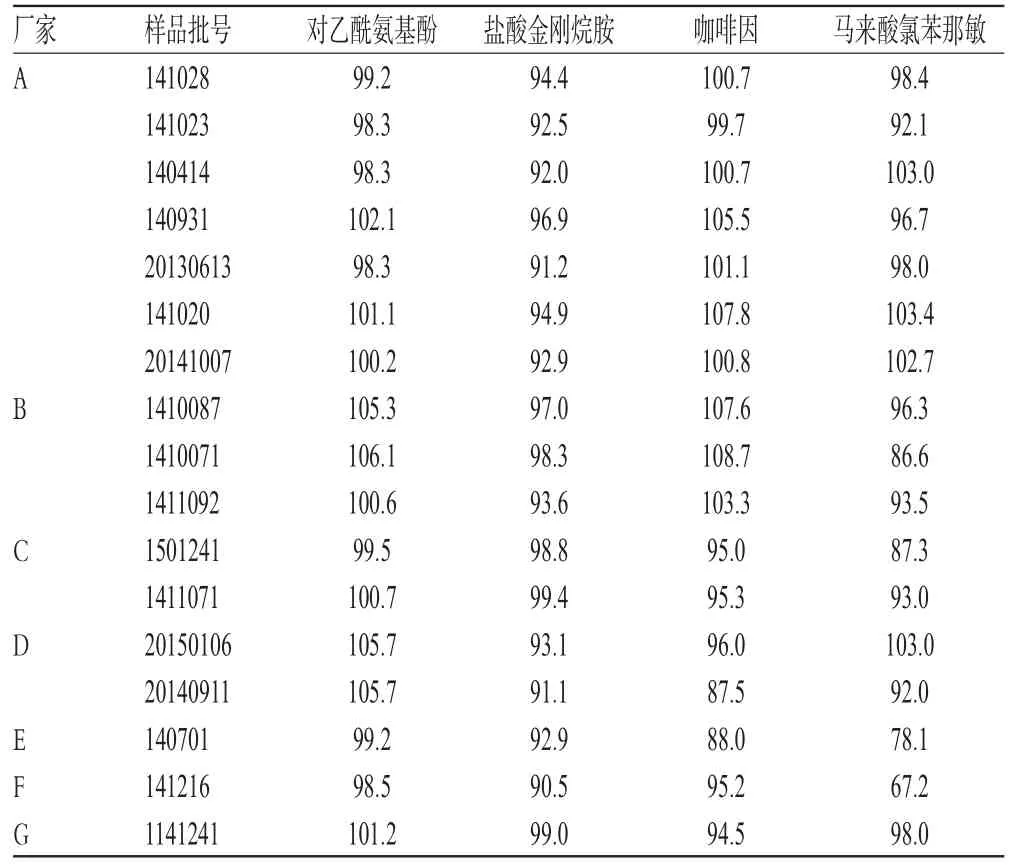
表2 樣品含量測定結果(n=3,%)Tab 2 Results of contents determination of samples(n=3,%)
3 討論
3.1 耐用性考察
本試驗分別選擇兩種色譜柱[HP-5石英毛細管柱(30 m×0.32 mm,0.25 μm),美國Agilent公司]、[DM-5石英毛細管柱(30 m×0.32 mm,0.25 μm),重慶迪馬工業有限責任公司],對對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的分離度進行考察。結果表明,不同品牌色譜柱對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的分離度影響不大,測定結果均符合要求,表明本方法的耐用性較好。
3.2 提取條件的選擇
本品為復方制劑,筆者參考相關文獻[8]并根據樣品及各成分的理化性質,選取了毒性較低的無水乙醇為溶劑,采用超聲振蕩15 min提取相關成分。結果,咖啡因、馬來酸氯苯那敏提取不完全;后改為80℃水浴加熱30 min進行提取,對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏均能提取完全。因此,本試驗采用80℃水浴加熱30 min進行提取。
綜上所述,本方法簡便快速、準確可靠,適用于復方氨酚烷胺片中對乙酰氨基酚、鹽酸金剛烷胺、咖啡因、馬來酸氯苯那敏的同時測定。
[1] 國家食品藥品監督管理局.復方氨酚烷胺片[S].WS1-XG-015-2002.2003.
[2] 國家食品藥品監督管理局.復方氨酚烷胺片[S].YBH06802005.2005.
[3] 田洪根,王洪明.HPLC法測定復方氨酚烷胺片中馬來酸氯苯那敏的含量及含量均勻度[J].中國藥品標準,2008,9(3):207-209.
[4] 石強.HPLC法同時測定復方氨酚烷胺片中咖啡因、馬來酸氯苯那敏的含量及含量均勻度[J].齊魯藥事,2012,31(10):585-587.
[5] 王軍,權勤波,宋建建,等.UFLC同時測定復方氨酚烷胺片中對乙酰氨基酚和咖啡因的含量[J].中國藥師,2009, 12(12):1775-1777.
[6] 謝華,傅萍,張悅楊,等.HPLC測定復方氨酚烷胺片中的對乙酰氨基酚、咖啡因和馬來酸氯苯那敏[J].華西藥學雜志,2012,27(3):307-308.
[7] 黃軍.RP-HPLC法測定復方氨酚烷胺片中4種主要成分的含量[J].生命科學儀器,2006,4(6):16-18.
[8] 黃義純,劉旺培.HPLC測定氨咖黃敏中對乙酰氨基酚和咖啡因的含量[J].首都醫藥,2007,14(16):472-474.
(編輯:劉 柳)
Simultaneous Determination of 4 Components in Compound Paracetamol and Amantadine Hydrochloride Tablet by GC
DONG Qiuxiang,ZHANG Yuehan,FU Pingping,LIU Zhiwei(Baoding Municipal Institute for Food and Drug Control,Hebei Baoding 071051,China)
OBJECTIVE:To establish a method for the simultaneous determination of paracetamol,amantadine hydrochloride,caffeine,chlorphenamine maleate in Compound paracetamol and amantadine hydrochloride tablet.METHODS:GC was performed on the column of HP-5 sillica capillary,temperature programmed,detector was FID detector,with the temperature of 300℃,carrier gas was nitrogen gas,the flow rate is 1.5 mL/min,the split ratio was 20∶1 and injection volume was 1 μL.RESULTS:The linear range was 156.0-4 990.4 μg/mL for paracetamol,125.7-4 023.2 μg/mL for amantadine hydrochloride,19.14-612.4 μg/mL for caffeine and 2.515-80.48 μg/mL for chlorphenamine maleate(all r=0.999 9);the limits of quantification were 1.4,0.5,1.1,0.9 ng,limits of detection were 0.4,0.2,0.3,0.3 ng;RSDs of precision,stability and reproducibility tests were lower than 2.0%;recoveries were 99.59%-101.77%(RSD=0.8%,n=9),99.56%-101.80%(RSD=0.7%,n=9),98.44%-100.83%(RSD=0.7%,n=9)and 100.05%-101.91%(RSD=0.6%,n=9),respectively.CONCLUSIONS:This method is simple,rapid,accurate and reliable,and suitable for the simultaneous determination of paracetamol,amantadine hydrochloride,caffeine,chlorphenamine maleate in Compound paracetamol and amantadine hydrochloride tablet.
Compound paracetamol and amantadine hydrochloride tablet;GC;Paracetamol;Amantadine hydrochloride;Caffeine;Chlorphenamine maleate;Content
R917
A
1001-0408(2017)06-0844-04
2016-02-28
2016-11-12)
*主管藥師,碩士。研究方向:化學藥品檢驗。電話:0312-5906322。E-mail:739998331@qq.com
DOI10.6039/j.issn.1001-0408.2017.06.34
