擬蛇算法在蛋白質折疊模擬中的應用可行性研究
2017-04-14 10:34:45張菁,郭丹,2
軟件 2017年2期
關鍵詞:研究
張 菁,郭 丹,2
擬蛇算法在蛋白質折疊模擬中的應用可行性研究
張 菁1,郭 丹1,2
(1. 哈爾濱工程大學,計算機科學與技術學院,黑龍江 哈爾濱 150001;2. 哈爾濱商業大學,計算機與信息工程學院,黑龍江 哈爾濱 150028)
本文以計算分子生物學中的蛋白質折疊模擬為研究對象,通過對一種新算法的可行性研究,意旨為領域內的算法研究提供一種可驗證性的算法。本文從收斂性和穩定性的角度,分析了擬蛇算法的核心函數,驗證該算法在蛋白質折疊模擬中的應用具有全局最優值的收斂性,并且進一步研究了這種仿生算法的運動穩定性,驗證了擬蛇算法的運動軌跡會達到一個穩定狀態,這與蛋白質折疊最后形成能量最小的穩定狀態的理論相吻合,驗證結果表明,擬蛇算法在蛋白質折疊模擬中應用具有可行性。
擬蛇算法;蛋白質折疊;可行性研究;擺
0 引言
計算分子生物學[1]是運用計算機軟件理論和技術,以分子生物學數據為研究對象的交叉研究領域。分子生物學的海量數據需要計算機學科的高效處理方法作為實驗前數據預處理、數據篩選、數據預測,以及實驗后的數據分析處理的重要技術支撐;另一方面,計算機對復雜大數據的處理研究,需要真實的有意義的訓練數據集,分子生物學的迅速發展,產生了大量的數據,而其間的關系也日趨復雜,這些龐大的復雜數據集合正是一個好的算法研究的數據驗證對象。
計算機模擬技術運用高速計算算法可以有效的對真實實驗中的復雜大數據集進行分析和處理,并且通過對不同實驗環境和實驗因素的計算機模擬可控性,得到更多實驗的可能性和更精確的理想化結論,也可以清晰的通過構象的變化來分析被模擬過程中哪些因素的影響格外重要,從而對功能關系的相互作用得出預測性的實驗依據幫助研究人員理解。
蛋白質折疊是分子生物學上生物自我組織的一個典型實證。計算分子生物學中的蛋白質折疊模擬算法是一個重要的研究對象。通過氨基酸長鏈彎曲、扭曲和折疊,蛋白質可以在三維結構上具有不同的構象。結構生物學已經驗證,蛋白質二維或三維結構決定了蛋白質的特定功能[2]。
目前人類對于折疊機制的了解程度還不足以精確的預測一個特定的序列如何折疊到天然構象,或用逆折疊方法設計出能折疊到具有穩定結構的真實蛋白的新序列[3]。雖然這種結構上的改變在自然界中只需要幾秒到幾分鐘,但到目前為止,即便是速度最快的計算機也無法完成。
蛋白質由數目龐大的氨基酸組成,氨基酸不同結構的排列可能性,決定了蛋白質構象可能具有無窮種結構性狀,這也就意味著模擬算法的機制盡可能的要接近真實折疊機制,在眾多預測算法中,很多仿生預測算法應運而生,用自然界已知的生物具有的自身特征性算法去解決未知的生物問題,如人工魚群算法[4,5]、遺傳算法[6,7]、人工神經網絡[8,9]、粒子群算法[10,11]、蟻群算法[12,13]等。擬蛇算法[14]是一種新穎的蛋白質折疊預測算法,本文對這一算法在蛋白質折疊模擬中的應用可行性進行研究,以推動該算法在領域研究中的運用。
1 擬蛇算法
自然界中,蛇不具有四肢,但它可以靠軀體的形狀的改變來獲得移動的動力。在這一點上,它與蛋白質無論從形狀到運動形式都有相似的地方。在粗糙得地面上蛇作一連串的波浪狀彎曲,體側不斷向地面施加壓力,由于地表的反作用力關系,所以能推動蛇體向前前進。軀體較粗的蛇或接近獵物時,常常采取直線運動方式爬行。這類蛇的特點是腹鱗與其下方的組織之間較疏松,由于肋骨與腹鱗間的肋皮肌有節奏的收縮,使寬大的腹鱗依次豎立于地面,于是蛇體就能不停地呈直線向前運動。捕捉到獵物后,蛇會將自身盤緊促使獵物窒息死亡。蛇的運動具有運動穩定性好,適應地形環境能力強等特點。
其運動具有以下特點:1)能在凹凸不平的地面上移動,有良好的地形自適應性;2)適合于在沼澤、沙漠等松軟環境中行進;3)能通過孔洞等狹小而又彎曲的通路,有較強的過障礙能力;4)能經常保持力學的穩定狀態。
蛋白質折疊的結果也是趨于形成低能量的力學的穩定狀態的物質。蛋白質的折疊過程經歷了一個中間狀態的,這種狀態被科學家形容為熔球[15]。熔球不像正常折疊后結構緊密地蛋白那樣脆弱,這樣的結構可以進行拉扯,不會被輕易破壞。熔球具有正常蛋白所有的二級結構,但是這種二級結構尚未正確折疊形成更高級的結構。當蛋白質折疊成穩定構型時,其能量是最低的。蛇的運動與蛋白質折疊在形狀和運動形式上有相似之處,因此研究和制作蛇形仿生算法來解決蛋白質折疊問題具有一定的研究可能性。
擬蛇算法作為一種仿生算法,主要以蛋白質折疊過程為研究對象,通過對蛇捕獲獵物的過程模擬蛋白質折疊過程。以蛇的三種運動模式:波浪運動、直線式運動和盤繞運動,去模擬蛋白質折疊過程:折疊初期的波浪運動、折疊過程中短暫的直線式運動和折疊后期的盤繞運動。
對于蛇捕食過程的描述:在其未發現捕食目標前的覓食過程中蛇做波浪運動,通過隨機探測目標的位置,即,當其與食物(外力)距離遠時(折疊開始時),或當區域空間大,擁擠濃度低時,蛇的軀體作波浪運動,其從頭部至尾部,形成若干個波峰和波谷,波峰和波谷隨時間交替改變,肽鏈的移動路徑是正弦波;一旦鎖定捕食目標,進入攻擊狀態,即,當其與食物(外力)距離近時(折疊中期),其改波浪運動為直線移動以快速接近獵物,長的肽鏈的移動路徑為直線式;當捕獲食物后(折疊后期),其做盤繞運動,將獵物盤緊以穩定捕獲狀態,進而完成整個捕食過程,肽鏈的移動路徑是螺旋曲線。
在蛋白質折疊模擬過程中擬蛇算法的實現步驟:1)初始狀態:隨機產生一條蛋白質序列的構型。給出氨基酸殘基個數,設定肽鏈折疊的最大距離參數,更新速度步長,給肽鏈最小能量設初值;2)當折疊距離小于折疊的最大距離時,做波浪運動,并計算肽鏈總的最小能量值;3)當折疊距離等于折疊的最大距離時,做直線運動,并計算肽鏈總的最小能量值;4)當折疊距離大于折疊的最大距離時,做盤繞運動,并計算肽鏈總的最小能量值。5)肽鏈總的最小能量不再變化時,輸出蛋白質構型,算法結束[16]。
擬蛇算法根據六個典型的蛋白質空間結構,在蛋白質折疊模擬過程中,通過改變參數演化兩種函數形式,圓型和扣型兩種類型。擬蛇算法的核心函數是:
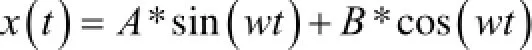
本文從收斂性和穩定性兩個方面,通過對核心函數的分析,驗證擬蛇在蛋白質折疊模擬中的應用可行性。
2 收斂性的分析
對于一種算法,其收斂性往往是人們所首要關心的問題。在擬蛇算法中,蛇的正弦移動行為奠定了算法收斂的基礎,直線移動行為增強了算法收斂的穩定性和全局性,盤繞行為則增強了算法收斂的快速性和全局性,其行為過程也對算法收斂的速度和穩定性提供了保障。總的來說,算法中對各參數的取值范圍還是很寬容的,并且對算法的初值也基本無要求。
擬蛇算法中殘基個數,殘基移動的最大速度和步長在迭代次數100時,這三個參數對算法收斂性的影響都是有限的,而合理選擇迭代次數是提高算法效率的關鍵。這一規律是否具有一般性還有待進一步研究。
由于算法參數不同,收斂過程和結果也存在一定的差異,所以在以下的討論中,將針對每一種參數連續多次進行全局尋優收斂實驗作為一組數據,然后對多組數據進行統計分析,從而確定各參數的性質。通常數據的均值(Mean)表征本組數據的優劣,其標準誤差(SE)則可以反映該組數據的穩定性。
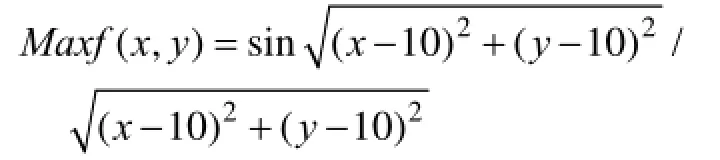
基于上面的二維正弦函數分析算法的收斂性,其中,0≤x≤20,0≤y≤20。該函數的全局極大值的周圍被無窮個極小值(波谷)所包圍,再外圍是無窮個局部極大值(波峰),在最遠端的四個角上又是處于局部極大值。使用該函數作為實驗函數應該具有一定的代表性。對于該函數,從擬蛇算法的任何初始值出發,都能收斂到全局最優值,并且收斂時間最短為幾秒。
3 穩定性分析
擬蛇算法的核心是一種振蕩形式[16]。一個理想的擺,力是Fkx=-,位置坐標x,質量為m。位置坐標可以表示為:x=x(t),速度函數為v=v(t)。這兩個函數彼此間有以下的關系:


方程式1定義速度函數,方程式2是牛頓定律用之于震蕩的擺,對式2兩邊對t求導,然后將式1帶入式2的求導,結果消去dx(t)/dt便得到一個二階常系數線性微分方程,可以解出v(t)
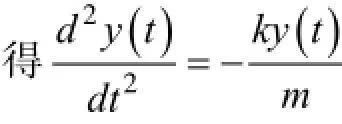
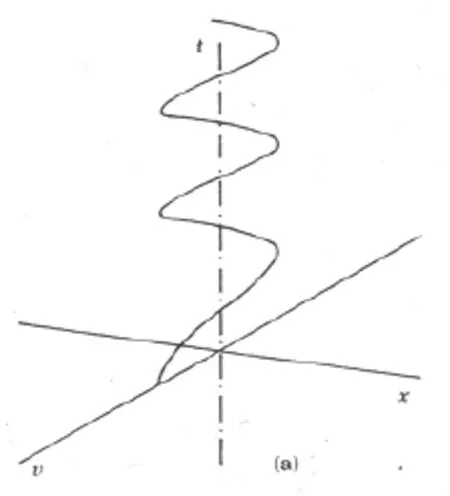
圖1a 速度v隨時間的t的變化Fig.1 a velocity change according to time
圖1給出由公式4所給出的擺狀態的時間演化圖。這個擺既在時間上有位置的移動,又在時間上有速度的變化,所以這系統的點的軌跡是上升的螺旋線。
相軌線[12-14]的概念在有關耦合非線性微分方程的討論中是很重要的,在缺乏完整分析一個動力學系統的情況下,擺的相軌線可用于有關運動本質的穩定性的分析。
相軌線是三維螺旋在v-x平面上的投影。也就是說我們要構造一個函數v=v(x)。這可以由公式5,6消去時間而做到。

所以我們可以得到:

對給出的初始條件來說,或者等價地說,對一個固定值K,公式(7)代表v-x平面上的一個橢圓如圖2所示。每一個K值,有一個橢圓與之對應。
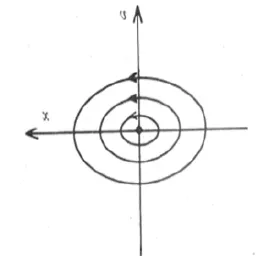
圖2 一個理想擺在相平面上的軌跡Fig.2 An idea Gradual stable focus
當相平面軌線的螺旋線趨向于奇點,如圖3,從而這個奇點叫做穩定焦點。在這種情況下,總的解趨向于一種振蕩運動中的定態,是漸近穩定的。
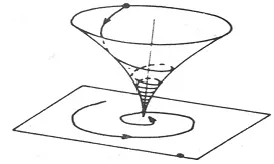
圖3 漸近穩定焦點,螺旋軌跡收斂于奇點Fig.3 Gradually to stable focus, screw track to odd spot
理想狀態下,擬蛇算法的核心函數在振蕩模型下,擺的特征方程:
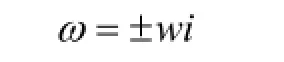
即iωω=±虛
擺有兩個純虛根,并且有,擺反映在坐標x與v上的代表點在相平面上隨時間而作周期的變化。因此,其相軌跡線必然都是一些圍繞奇點的封閉曲線。
擺是在小的瞬時的位移x=δx,擺就開始搖動,而此運動是x=x(t)與v=v(t)隨時間而減少,并最后為零,則擺的位置x=0,v=0,是一個穩定的狀態,我們把這一點叫做穩定奇點。
對于施加在擺上的每個初始力,會出現一條新的封閉的相軌線,如圖2所示。一個理想的擺可以規律性地振蕩著,只要沒有新的擾動去干擾它的運動,此時的奇點是穩定的。擺運動軌跡的穩定性恰好與蛋白質折疊最后形成能量最小的穩定狀態的理論相吻合。
4 結論
本文針對蛋白質折疊模擬研究中一種新算法的可行性研究,對擬蛇算法的核心函數進行了收斂性和穩定性分析。通過對算法收斂性的分析,以迭代次數100為限制,驗證該算法核心函數從任意初始值出發,都能收斂到全局最優值,并具有較快收斂速度,能有效地對蛋白質折疊進行模擬預測。通過對算法穩定性分析,從振蕩理論的擺軌跡的相平面穩定性研究,驗證了在理想狀態下擬蛇算法的運動軌跡在能達到一個穩定狀態,這與蛋白質折疊最后形成能量最小的穩定狀態的理論相吻合。擬蛇算法僅使用了目標問題的函數值,算法簡單,具有很強的跳出局部極值的能力,算法的穩定性確保其能快速跟蹤隨著工作狀況或其他因素的變更造成的極值點的漂移變化,獲得全局最優解。綜上所述,擬蛇算法在蛋白質折疊模擬研究中,具有應用可行性。
[1] ZHANG Y, Min X J. Computational molecular biology: an integration of experimental molecular and genome biology with computational technology: computational molecular biology[J]. 2011, 1(1): 1-3.
[2] GEORGINA M, DANCO D. Incorporating several features in the protein ray descriptor for more accurate protein 3D structure retrieval: Proceedings of the ACM workshop on 3Dobject retrieval [C]. 2012, 51-56.
[3] SCHAEFFER R D, DAGGETT V. Protein folds and protein folding: protein engineering design & selection[J]. 2011, 24(1-2): 11-19.
[4] CUI Z H, ZHANG Y. Swarm Intelligence in Bioinformatics: methods and implementations for discovering patterns of multiple sequences: journal of nanoscience and nanotechnology[J]. 2014, 14(2): 1746-1757.
[5] CUI Z H, ZHANG Y. Methods and implementations for discovering patterns of multiple sequences: journal of nanoscience and nanotechnology[J]. 2014, 14(2): 1746-1757.
[6] ISLAM M L, SHATABDA S, RAHMAN M S. GreMuTRRR: A novel genetic algorithm to solve distance geometry problem for protein structures: 2014 international conference on electrical and computer engineering (ICECE)[C]. 2014, 357-360.
[7] SHIN JM, LEE B, CHO KH. A new efficient conformational search method for ab initio protein folding study: window growth evolutionary algorithm: bulletin of the Korean chemical society[J]. 2016, 37(12) 1971-1976
[8] FLORIDO J P, POMARES H, ROJAS I. Prediction of functional associations between proteins by means of a cost-sensitive artificial neural network: lecture notes in computer science[J]. 2011, 6692: 194-201.
[9] THANGSUNAN P, KITTIWACHANA S, MEEPOWPAN P, KUNGWAN N, PRANGKIO P, HANNONGBUA S, SUREE N. Rapid activity prediction of HIV-1 integrase inhibitors: harnessing docking energetic components for empirical scoring by chemometric and artificial neural network approaches: journal of computer-aided molecular design [J]. 2016, 30(6): 471-488.
[10] KONDOV I. Protein structure prediction using distributed parallel particle swarm optimization : natural computing [J]. 2013, 12(1): 29-41.
[11] CHEUNG N J, DING X M,SHEN H B. Protein folds recognized by an intelligent predictor based-on evolutionary end structural information :journal of computational chemistry. 2016, 37(4): 426-436.
[12] OAKLEY M T,RICHARDSON E G, CARR H. Protein structure optimization with a "lamarckian" ant colony algorithm: IEEE-ACM transactions on computational biology and bioinformatics[C]. 2013, 10(6): 1548-1552.
[13] RAY S S,PAL S K. RNA Secondary structure prediction using soft computing: IEEE-ACM transactions on computational biology and bioinformatics[C]. 2013, 10(1): 2-17.
[14] 張天馳, 張菁. 模擬蛋白質折疊過程的新算法研究. 生物信息學[J]. 2011, 9(31): 142-145.
[15] https://en.wikipedia.org/wiki/Molten_globule
[16] 張菁, 張天馳. 蛋白質折疊過程模型. 應用科技[J]. 2011, 38(7): 41-43.
Application Feasibility Study on Snake Algorithm of Protein Folding Simulation
ZHANG Jing1, GUO Dan2
(1. Harbin Engineering University, College of Computer Science and Technology, Heilongjiang Harbin 150001, China; 2. Harbin University of Commerce, School of Computer and Information Engineering, Heilongjiang Harbin 150028, China)
The research object of the paper is a new simulation algorithm for protein folding in computational molecular biology. From the view of the astringency and stability, the algorithm research in the field of the purpose of this paper is to provide a kind of algorithm of verifiability. In this paper, snake algorithm was validated is accord with the global optimum of the astringency in the application of protein folding simulation by analyses the core function. And the project further studies the movement stability of the bionic algorithm. Proved that the proposed algorithm of snake trajectory will reach a steady state, which was consistent with Minimum energy of the formation of stable state theory when protein folding is complete. Results show that the proposed snake algorithm was feasible for application in the numerical simulation of protein folding.
Snake algorithm; Protein folding; Feasibility study; Pendulum
TP39 3.08
A
10.3969/j.issn.1003-6970.2017.02.017
黑龍江省教育廳科學技術研究基金項目(12541211)
張菁(1965-),女,博士后,教授,博士生導師,研究方向為計算分子生物學,虛擬現實,醫學圖像處理;郭丹(1978-),女,博士研究生,副教授,研究方向為計算分子生物學,大數據可視化。
本文著錄格式:張菁,郭丹. 擬蛇算法在蛋白質折疊模擬中的應用可行性研究[J]. 軟件,2017,38(2):75-79
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
