19例線粒體腦肌病的電子顯微鏡研究
2017-05-20 07:44:45孫異臨張在強李少武王芹芹張翠萍皇埔秉軒
分析儀器 2017年2期
陳 晨 孫異臨 張在強 李少武 張 巖 王芹芹 張翠萍 皇埔秉軒
(1.首都醫科大學 北京市神經外科研究所,北京100050;2.陸軍總醫院 八一腦科醫院,北京100007)
19例線粒體腦肌病的電子顯微鏡研究
陳 晨2孫異臨1*張在強1李少武1張 巖2王芹芹2張翠萍1皇埔秉軒2
(1.首都醫科大學 北京市神經外科研究所,北京100050;2.陸軍總醫院 八一腦科醫院,北京100007)
目的:探討線粒體腦肌病的超微病理特點和發病機制。線粒體腦肌病MELAS (mitochondrial encephal myopathy with lactic acidosis and strokelike episodes)是一種以卒中樣發作同時伴有血乳酸增高為主要臨床表現的線粒體病,病因為遺傳性基因缺陷所致。方法:對19例線粒體腦肌病患者肌肉活檢標本的超微病理變化及核磁共振特點進行分析。結果:研究提示核磁共振檢查MELAS病患者大腦顳、頂、枕皮層可見不按血管解剖學分布的梗死樣病灶;電子顯微鏡檢查見病變肌細胞內線粒體形態異常,出現矩陣樣結晶包涵體結構。結論:MELAS的診斷需結合臨床、病理、電鏡、影像、血清學和基因檢查,其中肌肉活檢的電子顯微鏡檢查可作為金標準。
線粒體腦肌病 超微病理 電子顯微鏡
線粒體腦肌病(MELAS)多發生在青少年,是以腦和肌肉受累為主的多系統病變,臨床癥狀為全身性表現,可以有頭痛、嘔吐、發作性癲癇,此外還可見卒中樣發作,患者智力低下、生長緩慢、身材矮小、肌肉無力、骨骼肌極度不能耐受疲勞、視聽能力下降等,血清學檢查可見血乳酸水平明顯升高,肌酶升高〔1〕。
1 臨床資料和方法
本組共19個病例,其中男性16例,女性3例,青少年發病,臨床癥狀表現為卒中樣發作,血清學檢查均可見血乳酸水平升高。其他癥狀和體征有頭痛、發作性癲癇,智力低下、身材矮小、骨骼肌不能耐受疲勞、聽力下降等。
標本制備:送檢標本為患者腓腸肌,于手術臺邊取材新鮮標本即刻置入2%多聚甲醛-2.5%戊二醛固定液內進行初固定,0.1M二甲砷酸鈉緩沖液(pH7.4)沖洗后用Leica自動組織處理機處理標本,先后經1%四氧化鋨后固定,雙蒸水沖洗,梯度乙醇脫水至環氧丙烷,SPI812浸透包埋;制備1μ半薄切片,天青-美藍染色,光學顯微鏡檢查;光鏡定位后用Leica UC6型超薄切片機制備40納米厚度超薄切片,經醋酸雙氧鈾、檸檬酸鉛染色,在日立H-7650透射電子顯微鏡觀察及Gatan 832CCD相機拍照。
2 檢查結果
神經影像學MRI檢查表現為大腦后半部的顳、頂、枕皮層及白質區見梗死樣病灶,可見顳頂枕皮層多發層狀長T1、T2異常信號,梗死樣病灶特點為不按血管的解剖學分布,即病灶與腦動脈供血分布區域不一致,梗死灶周圍無強化(圖1)。
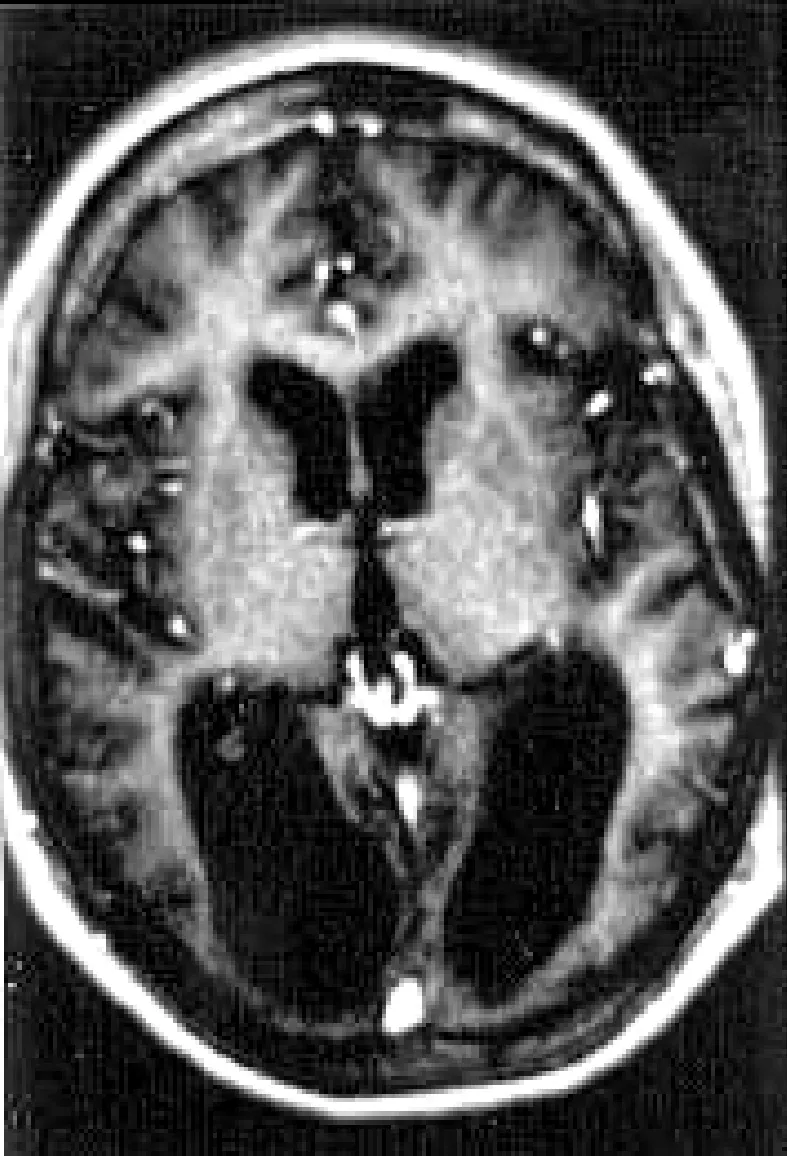
圖1 MRI顳頂枕皮層多發異常信號,梗死樣病灶不按血管的解剖學分布
病理學檢查可見病變肌細胞與正常肌細胞混合鑲嵌在一起,病變細胞在改良Gomori染色時出現不規則的紅色邊緣(ragged-red fibers RRF)(圖2);還原型輔酶Ⅰ四唑氮還原酶(NADH)染色可見肌膜下深染(圖3);PAS及油紅O染色可見糖原和脂質沉積。
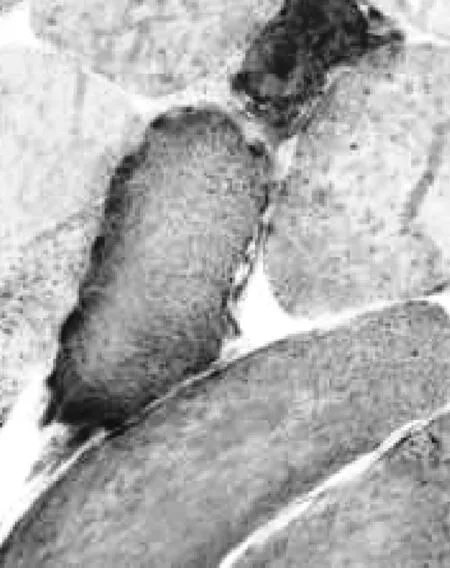
圖2 改良Gomori染色時出現不規則的紅色邊緣
電子顯微鏡檢查見病變肌細胞的肌膜下和肌原纖維之間線粒體明顯增多且形態異常(圖4)、線粒體內矩陣樣結晶包涵體的結構變化多樣,可見不規則的幾何圖形線粒體、也有的呈規則的晶體樣結構或呈同心圓狀(圖5);部分病例除病變肌細胞內線粒體數量及形態異常外,同時可伴有糖原顆粒及脂類物質聚集在肌絲之間(圖6)。

圖4 病變肌細胞的肌膜下和肌原纖維之間線粒體明顯增多
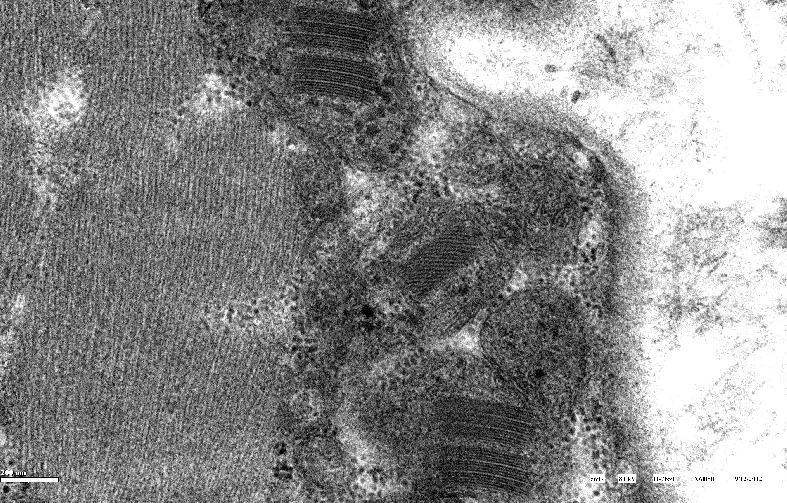
圖5 病變肌細胞內見不規則幾何圖形線粒體、有的呈規則的晶體樣結構
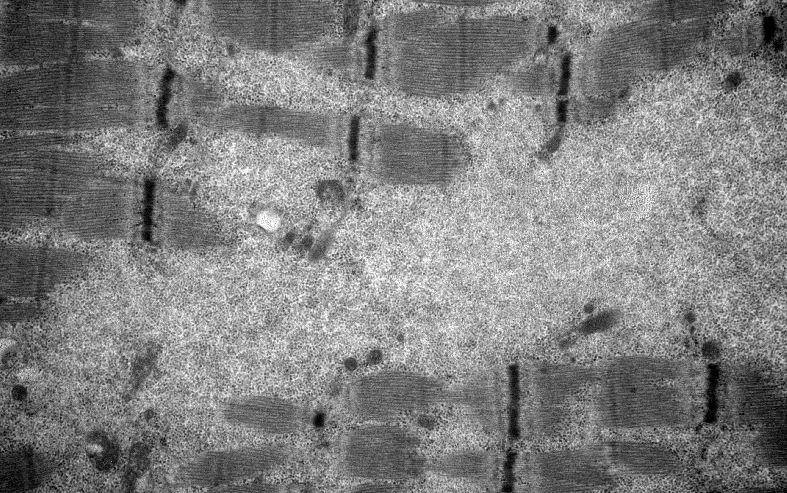
圖6 肌細胞內肌絲斷裂,線粒體形態異常,同時伴有糖原顆粒蓄積
3 討論
MELAS的腦卒中樣發作可累及大腦后半部的顳、頂、枕皮層及白質區,病變范圍較廣且具有一定的對稱性,梗死灶周圍無強化無明顯水腫。CT及核磁共振等神經影像學檢查對線粒體腦肌病診斷具有重要輔助作用,常可見到蒼白球、丘腦、基底節鈣化,腦室周圍白質病變以及不按血管解剖學分布的多發腦梗塞。如進行MR波譜成像檢查,磁共振波譜可以檢測組織內能量代謝和生化的改變,顯示腦內病灶中乳酸含量可進一步明確診斷。本組病例MELAS患者MRI檢查可見顳頂枕皮層多發層狀長T1,T2異常信號,病灶特點不按血管的解剖學分布即梗死樣病灶與腦動脈供血分布區域不一致,病灶處血管分支少,血流量低下,這一點與缺血性腦梗死病灶的分布不同,說明線粒體腦肌病是線粒體功能異常引起的腦內微小血管病變,不累及顱內大的血管,所以是代謝性腦梗塞〔2〕。腦電圖檢查在伴有癲癇發作的線粒體腦肌病患者有一定意義,以皮層損害為主的病變表現為彌散性全腦的異常腦電波;部分病例也可見局灶性的改變,出現癲癇特有的棘慢波;肌電圖檢查多表現為肌源性損害改變,也有部分患者同時出現肌源性或和神經源性損害改變。由于線粒體功能異常ATP合成受阻,無氧糖酵解增多,因而出現相應的臨床癥狀,如肌無力及不能耐受疲勞等〔3〕,患者血清學檢查乳酸升高是一項重要的診斷指標。
線粒體腦肌病的病理學檢查是診斷該病的必要手段,肌肉組織活檢可見呼吸鏈酶缺失的病變肌細胞與正常骨骼肌細胞混合鑲嵌在一起,這種線粒體異常增多的病變肌細胞在改良Gomori染色時肌細胞出現不規則的紅色邊緣,這種紅色邊緣肌纖維(ragged-red fibers RRF)是由于ATP合成不足肌細胞為代償而產生的肌酸激酶沉積所致;還原型輔酶Ⅰ四唑氮還原酶(NADH)染色可見到肌膜下深染的肌細胞;PAS及油紅O染色可見糖原和脂質沉積,同時可見病變肌細胞變性、大小不等及細胞核內移的現象〔4〕。
電子顯微鏡檢查見病變肌細胞的超微結構變化復雜,肌膜下和肌原纖維之間線粒體數量明顯增多且形態異常、線粒體內矩陣樣結晶包涵體的結構變化多樣〔5〕,有些為不規則的幾何圖形、也有的呈高度規則的晶體、或呈周期性編織樣結構也可以呈同心圓狀〔6〕,這些結構的產生是線粒體功能損害的結果還是導致線粒體功能喪失的原因尚不清楚。線粒體內不同類型的結晶樣包涵體可能與不同性質的蛋白質聚集有關,也可能是同一種蛋白在不同病變階段的異常合成所致〔7〕。線粒體在糖原、脂肪代謝過程中關系密切,由于線粒體功能異常能量代謝障礙,常常累及糖原和脂質的代謝異常,因此部分病例在病變肌細胞內線粒體變化的同時可伴有糖原顆粒及脂類物質聚集,也可見較多脂褐素,這些糖原和脂類物質的積聚均繼發于線粒體功能的障礙,同時過多的脂肪累積反過來又抑制線粒體內酶的功能〔8〕,肌細胞內脂肪代謝障礙造成的脂質堆積是引起肌肉極度不能耐受疲勞的原因之一。
線粒體是與機體內能量代謝相關的細胞器,即細胞內氧化磷酸化的場所。在線粒體的遺傳學研究中已知,線粒體具有母系遺傳的獨立基因即線粒體DNA(Mitochondrial DAN, mtDNA),線粒體DNA是人體細胞中唯一的核外DNA, mtDAN編碼的蛋白質只有十幾種,主要是細胞色素b、細胞色素c、NADH脫氫酶和氧化酶的亞單位,具有全套的呼吸鏈酶體系,其功能主要是氧化底物,利用呼吸鏈電子傳遞合成的ATP。Goto報道MELAS的基因突變位點主要為線粒體tRNA亮氨酸基因的A3243G核苷酸的點突變,由A變為G是導致線粒體腦肌病的主要致病因素,少部分是T3271C位置的點突變〔9〕。這種遺傳缺陷導致線粒體蛋白質翻譯、合成錯誤,引起線粒體呼吸鏈受損,造成線粒體酶系缺乏以致氧化磷酸化異常,最終引起線粒體腦肌病的發生。在線粒體功能發生障礙時,三磷酸腺苷ATP生成減少,導致ATP閾值較高的器官如腦組織、骨骼肌和心肌等高度依賴氧化磷酸化代謝的高耗氧量組織出現功能衰竭,引起全身多器官受累,當引起中樞神經系統發生病變時即為線粒體腦肌病MELAS。此外由于mtDNA不與組蛋白結合,因此其編碼的蛋白質容易受到活性氧自由基的損害,當mtDNA損害積累增多時就會造成線粒體結構和功能異常,繼而引起線粒體病。另外由于mtDNA沒有修復系統,線粒體DNA的突變、重復、缺失即可導致線粒體病的發生,同時線粒體還參與活性氧產生和誘導凋亡的病理過程,因此當線粒體內膜上細胞色素c激活caspase蛋白通道時即可導致細胞凋亡。
臨床工作如見到青少年腦卒中樣發作,同時伴有高乳酸血癥的患者,應高度警惕線粒體腦肌病。患者的腦卒中樣發作是由于局部腦組織ATP合成減少所致,并不是缺血性梗死。患者高乳酸血癥是由于線粒體病變導致ATP生成減少,為代償氧化磷酸化不足,組織內糖解酵代謝亢進,引起的血清內乳酸蓄積。
研究結果提示:(1)MELAS的診斷需結合臨床、病理、電鏡、影像、血清學和基因檢查,其中肌肉活檢的電鏡診斷最為重要,因為電鏡檢查可以得到具有決定性診斷意義的形態學證據,因此這項檢查是診斷線粒體病的金標準;(2)MRI證實線粒體腦肌病引起的卒中樣發作是線粒體功能異常引起的腦內微小血管病變,不累及顱內大的血管;(3)患者血清內乳酸升高是由于線粒體病變導致三磷酸腺苷ATP生成減少,組織內糖解酵代謝亢進所致,是引起肌無力和肌肉疼痛的原因之一;(4)線粒體功能異常累及糖原和脂質的代謝異常,引起糖原和脂類物質的積聚是引起肌肉極度不能耐受疲勞的原因。
[1]Kobayashi Y, Momoi MY, Tominaga K, et al . Respiration deficient cells are caused by a single point mutation in mitochondrial tNRA-Leu (UUR) gene in mitochondrial myopathy, encephalopathy, lactic acidosis and strokelike episodes(MELAS)〔J〕.Am J Hum Genet,1991,49:590-599.
[2] 盧巖,王向波.線粒體腦肌病MELAS型的影像學和病理學特征〔J〕.中國腦血管病雜志, 2006,3(2):81-86.
[3] Kaufinann P, Shungu D C, Sano M C, et al. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology ,2004,62(8):1297-1302.
[4]陳清棠, 李曉東.原發性線粒體肌病和腦肌病〔J〕. 臨床神經病學雜志,2003,16(4):249-250.
[5] 陳莉,盧德宏,徐慶中. 線粒體腦肌病的神經病理學〔J〕. 卒中與神經疾病雜志, 1998,5(3):173-175.
[6] Ban S, Mori N, Saito K, Mizukami K, et al. Shiraishi H, An autopsy case of mitochondrial encephalomyopathy (MELAS) with special reference to extra-neuromuscular abnormalitie〔J〕.Acta pathol Jpn,1992,42(11):818-825.
[7] Farrants G W, Hovmoller S, Stadhouders A M, Two type of mitochondrial crystals in diseased human skeletal muscle fibers〔J〕.Muscle and Nerve 1988,11(1):45-55.
[8] Campos Y, Huertas R, Bautista J, Muscle carnitine deficiency and lipid storge myopathy in patients with mitoychondrial myopathy 〔J〕.Muscle and Nerve, 1993,16:778-781.
[9] Goto Y I, Nonaka I A, Mutation in the tRNA Leu(UUR) gene assotiated with the MELAS subgroup of the mitochondrial encephalomyopaties 〔J〕.Nature 1990,348:651-656.
TEM investigation on ultrastructural pathology of mitochondrial encephal myopathy.
Chen Chen2, Sun Yilin1*Zhang Zaiqiang1, Li Shaowu1, Zhang Yan2, Wang Qinqin2, Zhang Cuiping1, Huangpu Bingxuan2
(1.Beijing Neurosurgical Institute,Capital Medical University,Beijing 100050,China; 2.Brain Hospital,Army General Hospital , Beijing 100007,China))
In this paper, ultrastructural pathological change and MRI feature of muscle biopsy specimens of mitochondrial myopathy in 19 cases were analyzed. MRI examination indicated that there were infarct-like lesions in cerebral temporal, top and occipital cortex, the vascular distribution was not according to anatomy. Electron microscopy showed abnormal mitochondrial morphology changes in muscle cells and matrix like crystalline inclusions structure. MELAS diagnosis should be combined with clinic, pathology, electron microscopy, imaging, serology and genetic examination, of which the electron microscopic diagnosis of the muscle biopsy is the most important.
mitochondrial encephalo myopathy;ultrapathology;electron microscopy
陳晨,女,1988年出生,畢業于內蒙古醫科大學,大學本科,工作單位:陸軍總醫院附屬八一腦科醫院電鏡室。
*通訊作者:孫異臨 ,男,1952年出生, 研究員,E-mail:13020068718@163.com。
10.3936/j.issn.1001-232x.2017.02.013
2016-12-06
