SPE-HPLC-MS/MS檢測動物源性食品中金剛烷胺殘留
2017-11-10 22:03:40云雅光池永紅王碩
食品研究與開發 2017年22期
關鍵詞:檢測
云雅光,池永紅,王碩
(1.天津科技大學,天津300457;2.包頭輕工職業技術學院,內蒙古包頭014035)
SPE-HPLC-MS/MS檢測動物源性食品中金剛烷胺殘留
云雅光1,2,池永紅2,王碩1,*
(1.天津科技大學,天津300457;2.包頭輕工職業技術學院,內蒙古包頭014035)
建立一種固相萃取-高效液相色譜-串聯質譜(Solid phase extraction coupled to high performance liquid chromatography tandem mass spectrometry,SPE-HPLC-MS/MS)檢測動物源性食品中金剛烷胺殘留的方法。研究采用Oasis MCX固相萃取小柱為基質凈化柱,以Agilent SB-C18柱(2.1 mm×150 mm,3.5 μm)為液相分離柱,0.1%甲酸-乙腈為(體積比,80∶20)為流動相,流速0.2 mL/min。用電噴霧離子源正離子多反應監測(Multi-reaction monitoring,MRM)模式進行檢測,外標法定量。在0.1 μg/L~100.0 μg/L范圍內具有較好的線性關系,相關系數>0.999。該方法檢出限(Limit of detection,LOD)為 1.0 μg/kg,定量限(Limit of quantitation,LOQ)為 3.0 μg/kg,對 3 個添加濃度(30.0、60.0、90.0 μg/kg)下的雞胸,雞肝,雞蛋,豬肉,羊肉5種樣品中金剛烷胺殘留的檢測具有較高的準確度(回收率在83.6%~94.2%之間)和重現性(RSD<4.0%,n=3)。
金剛烷胺;高效液相色譜串聯質譜;固相萃取;動物源性食品;檢測
金剛烷胺是人工合成飽和三環癸烷的氨基衍生物,用于亞洲A型流感病毒的預防和早期治療,也可用于治療帕金森病引起的神經障礙和預防動物常見的病毒類疾病如禽流感等[1-4]。由于價格低廉,曾廣泛運用于禽獸養殖業,長期使用會形成藥物殘留,從而導致抗藥性[5],神經過敏,焦慮,夢魘,間歇性的幻覺等[6]一系列的負作用。我國于2005年12月發布《關于清查金剛烷胺等抗病毒藥物的緊急通知》,禁止金剛烷胺等抗病毒藥作為獸用藥[7],FDA規定禁止將人類抗病毒藥物用于畜禽類的防治[8]。因此,為了減少對人類的潛在危害,建立一種簡單,廉價,高靈敏性的方法來檢測動物源性食品中金剛烷胺的殘留是非常必要的。
固相萃取(Solid-Phase Extraction,SPE)是近年發展起來一種樣品預處理技術,由液固萃取和柱液相色譜技術相結合發展而來,主要用于樣品的分離、純化和富集,目的在于降低樣品基質干擾,提高檢測靈敏度。與傳統的液液萃取法相比較可以提高分析物的回收率,更有效的將分析物與干擾組分分離,減少樣品預處理過程,操作簡單、省時、省力。廣泛的應用在食品中農獸藥及食品添加劑殘留、環境及藥物代謝等領域的檢測。
目前公開發表的檢測金剛烷胺殘留的文獻方法主要有氣相色譜法[9-11]、高效液相色譜[12-15]、高效液相色譜串聯質譜[16-19],分光光度法[20-22]和電位滴定法[23-24]。HPLC-MS/MS法因具有高效分離和集組分定性、定量于一體等優異性能,成為近年來獸藥殘留檢測方法研究的主要方向。本研究采用固相萃取結合高效液相色譜-串聯質譜技術,分別對樣品前處理條件(提取劑,固相萃取小柱)及金剛烷胺質譜條件進行了優化,建立了一種簡單、快速、準確測定動物源性食品中金剛烷胺殘留的方法。
1 材料與方法
1.1 主要儀器與試劑
Agilent LC-1200,Agilent 6410,Triple Quad LC/MS高效液相色譜串聯質譜體系:美國Agilent公司;AM-6勻漿組織破碎機:日本NISSEI公司;Centrifuge5804R臺式冷凍離心機:德國Eppendorf公司;QL-901振蕩器:海門其林貝爾儀器制造有限公司;Oasis MCX,Oasis PCX,Oasis MAX 固相萃取柱:3 cc,60 mg,Waters公司;超純水系統(18.2 MΩ cm):美國Labconco公司。
金剛烷胺標準品:美國Sigma-Aldrich公司;甲醇、乙醇、乙腈(色譜純)有機溶劑:天津化學試劑一廠。雞胸、雞肝、雞蛋、豬肉、羊肉:天津某超市。
1.2 液相色譜及質譜條件
高效液相色譜串聯質譜體系(Agilent LC-1200;Agilent 6410,Triple Quad LC/MS;USA)。
色譜條件:色譜柱為Agilent SB-C18柱(2.1 mm×150 mm,3.5 μm;),流速為0.2 mL/min,柱溫為25℃。流動相由乙腈和0.1%甲酸的混合溶液(體積比:20/80)組成。進樣量為1 μL。此外,流動相在使用前需要經過0.22 μm濾膜進行過濾處理。
質譜條件:電噴霧離子源(ESI),干燥器溫度350℃,霧化氣為N2,流速10 L/min,霧化氣壓力40 psi(1 psi=6.895×103Pa),毛細管電壓 4.0 kV,碰撞氣為 N2,多反應檢測模式(MRM)。金剛烷胺的質譜圖見圖1。
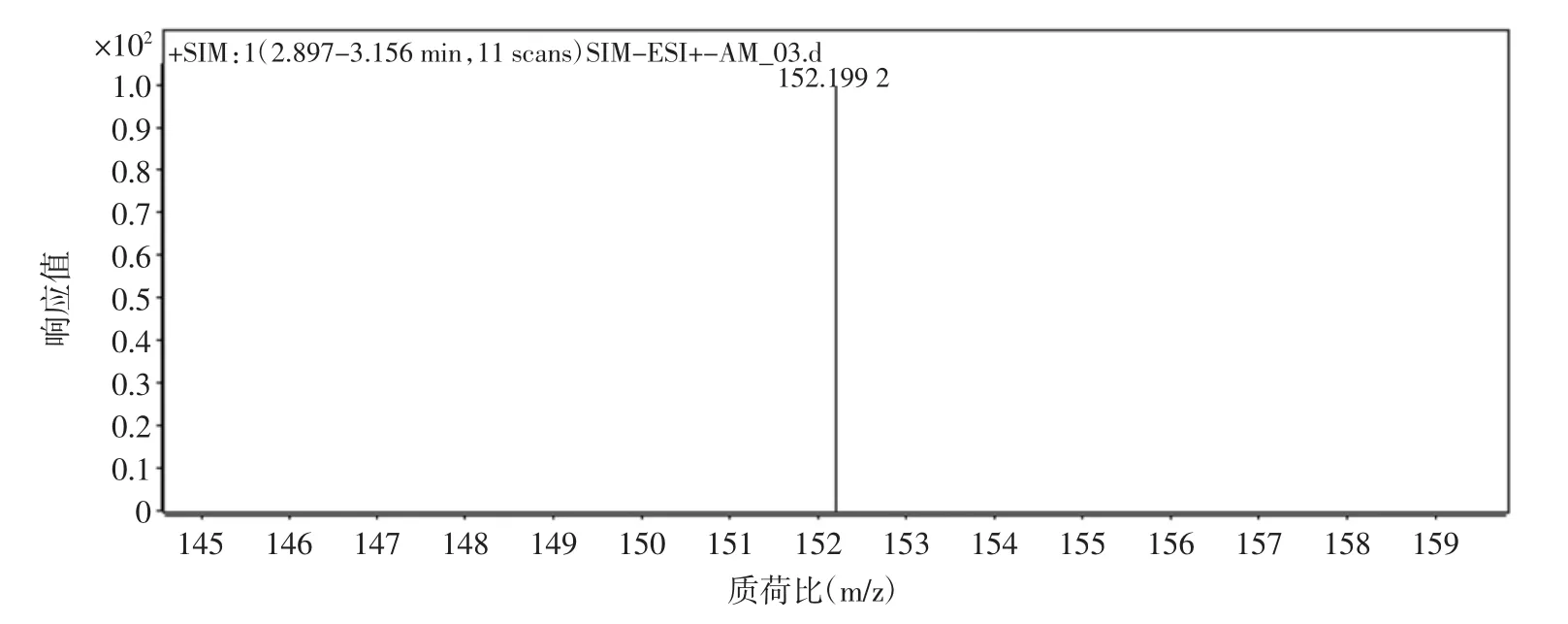
圖1 金剛烷胺[M+H]+的分子離子質譜圖Fig.1 Product ion mass spectrum diagram of amantadine
1.3 標準溶液的配制
準確稱金剛烷胺標準品0.010 g,溶解于10 mL甲醇中,并定容到100 mL,搖勻即得100.0 mg/L標準品儲備液,4℃冷藏,儲存期一個月。
準確量取金剛烷胺標準品儲備液1.0 mL,用甲醇稀釋定容至100 mL,即得1.0 mg/L標準工作液。取8個10 mL容量瓶,分別加入標準工作液1.0、5.0、10.0、50.0、100.0、250.0、500.0、1 000 μL,加甲醇定容至刻度,搖勻,即得到濃度為 0.1、0.5、1.0、5.0、10.0、25.0、50.0、100.0 μg/L 系列標準溶液。
1.4 樣品前處理
本研究選用5種不同的樣品基質(雞胸,雞肝,雞蛋,豬肉,羊肉)均購于天津本地某超市。
樣品預處理過程如下:準確稱取2.00 g(精確到0.01 g)粉碎的樣品,置于50 mL離心管中,加入10 mL乙腈-1%三氯乙酸(體積比,1∶1)溶液。然后將獲得的混合物渦旋2 min,超聲30 min,在-4℃下10 000 r/min離心10 min。然后將上清液轉移到試管中用于固相萃取凈化。
凈化過程如下:先用3.0 mL甲醇和3.0 mL雙蒸水活化萃取柱,流速均為15 mL/min。取5.0 mL提取液上柱,流速為1.0 mL/min。依次用2%鹽酸和甲醇各3.0 mL來淋洗,流速為1.0 mL/min。然后用5.0 mL氨水-甲醇-異丙醇(體積比,5∶90∶5)來洗脫,流速為1.0 mL/min。將洗滌液轉移到試管中,氮氣吹干。獲得的殘渣用1.0 mL甲醇進行復溶,再經0.22 μm濾膜過濾后用于高效液相色譜串聯質譜方法分析。
2 結果與分析
2.1 前處理條件的優化
對濃度為1.0 μg/L的金剛烷胺標準溶液,在相同的實驗條件下,分別使用甲醇-1%三氯乙酸(體積比,1∶1)、乙腈-1%三氯乙酸(體積比,1∶1)、1%乙酸乙腈、1%甲酸乙腈、甲醇和乙腈作為提取液,通過回收率來對比提取效果,結果如圖2。
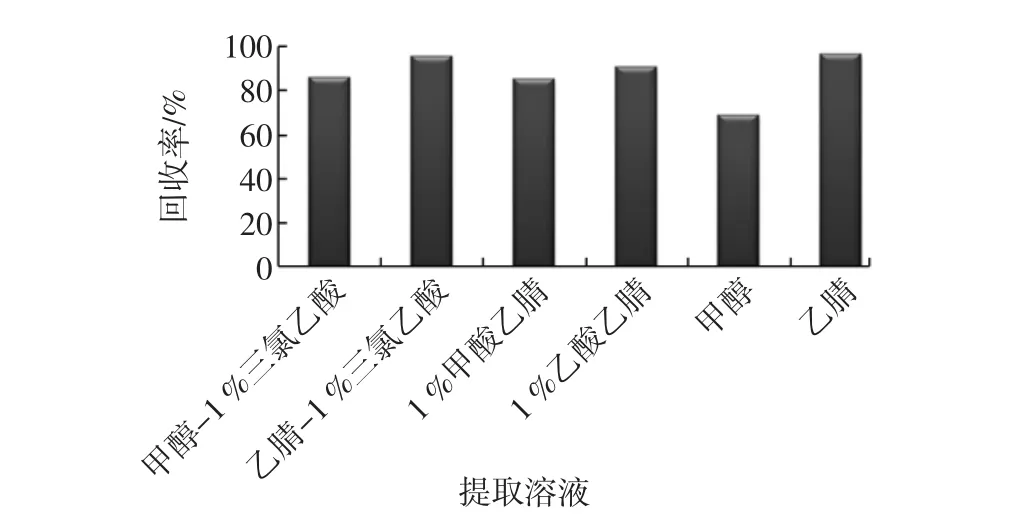
圖2 不同提取溶劑對金剛烷胺提取效果的影響(n=3)Fig.2 The influence of different extraction solvents on the amantadine extraction efficiency(n=3)
可見,采用乙腈-1%三氯乙酸(體積比,1∶1)和純乙腈的提取的回收率最高,但考慮到實際樣品中存在蛋白及脂肪,因而本方法選擇乙腈-1%三氯乙酸(體積比,1 ∶1)作為提取液。
試驗采用空白樣品加標的方式,比較了WatersOasis PCX(3 mL,60 mg)、Waters Oasis MCX(3 mL,60 mg)、Waters Oasis MAX(3 mL,60 mg)3種固相萃取柱的凈化效果,結果表明,在 0.5,2.0,20.0 μg/kg 3 個加標水平下,Waters Oasis PCX、Waters Oasis MAX小柱的平均回收率分別為83.7%和73.0%,而Oasis MCX的平均回收率均在96.5%以上,試驗結果見表1。最終試驗選擇采用Waters Oasis MCX(3 mL,60 mg)固相萃取小柱凈化樣品。

表1 金剛烷胺回收率和相對標準偏差(RSD)(n=3)Table 1 Recoveries and RSDs of amantadine standard with different SPE column(n=3)
2.2 質譜條件優化與選擇
金剛烷胺鹽酸鹽溶于水后,極易得到H+,形成穩定的[M+H]+準分子離子,選擇正離子電離(ESI+)方式,對金剛烷胺進行質譜條件的優化。先對金剛烷胺進行一級質譜掃描,得到相應的母離子峰;再對其進行二級質譜掃描,得到子離子信息。然后,在選擇多反應監測(MRM)模式下對得到離子對進行質譜參數優化。試驗結果表明,在電噴霧正離子模式下,金剛烷胺可以生成[M+H]+(m/z152.1)分子離子峰,試驗對噴霧電壓,錐孔電壓,碰撞電壓等質譜條件進行優化,得到定性離子對 152.1/135.1,152.1/93.1,152.1/79.1 效果較好,試驗結果見表2。金剛烷胺的二級質譜圖見圖3。

表2 金剛烷胺的質譜參數Table 2 Mass spectrometer conditions of Amantadine
2.3 標準曲線的繪制及實際樣品檢測
按照1.3所示方法配制不同濃度(X,μg/L)的金剛烷胺分析物的標準工作液,并按試驗所列色譜及質譜條件進行分析測定。在0.1 μg/L~100.0 μg/L范圍內呈良好線性關系(R2>0.999),標準曲線分別為:Y=1.021X+0.111。試驗數據確定本方法的定量限(LOQ)為 3.0 μg/kg。
研究中以雞胸,雞肝,雞蛋,豬肉,羊肉為代表的動物源性食品進行添加回收試驗,以考察所建立的基質凈化方法及金剛烷胺檢測方法的實際應用能力。準確稱取的雞胸,雞肝,雞蛋,豬肉,羊肉樣品各2.0 g,分別進行金剛烷胺濃度為 30.0、60.0、90.0 μg/kg的濃度添加,每個濃度平行3個,樣品前處理過程如上所述。基質提取液上機分析取峰面積根據標準曲線計算目標分析物的濃度,以加入量與測得量之比計算回收率及相對標準偏差。結果如表3所示。
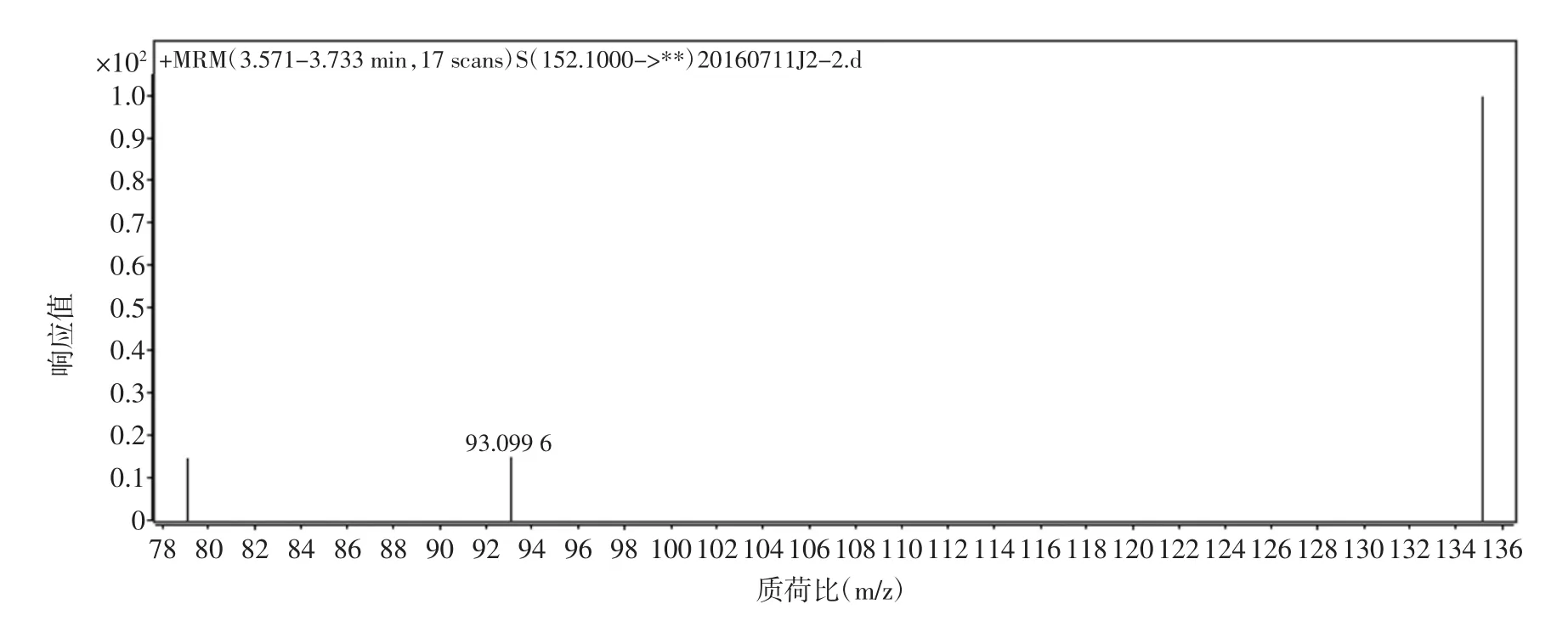
圖3 金剛烷胺的二級質譜圖Fig.3 Product ion mass spectrum diagram of amantadine
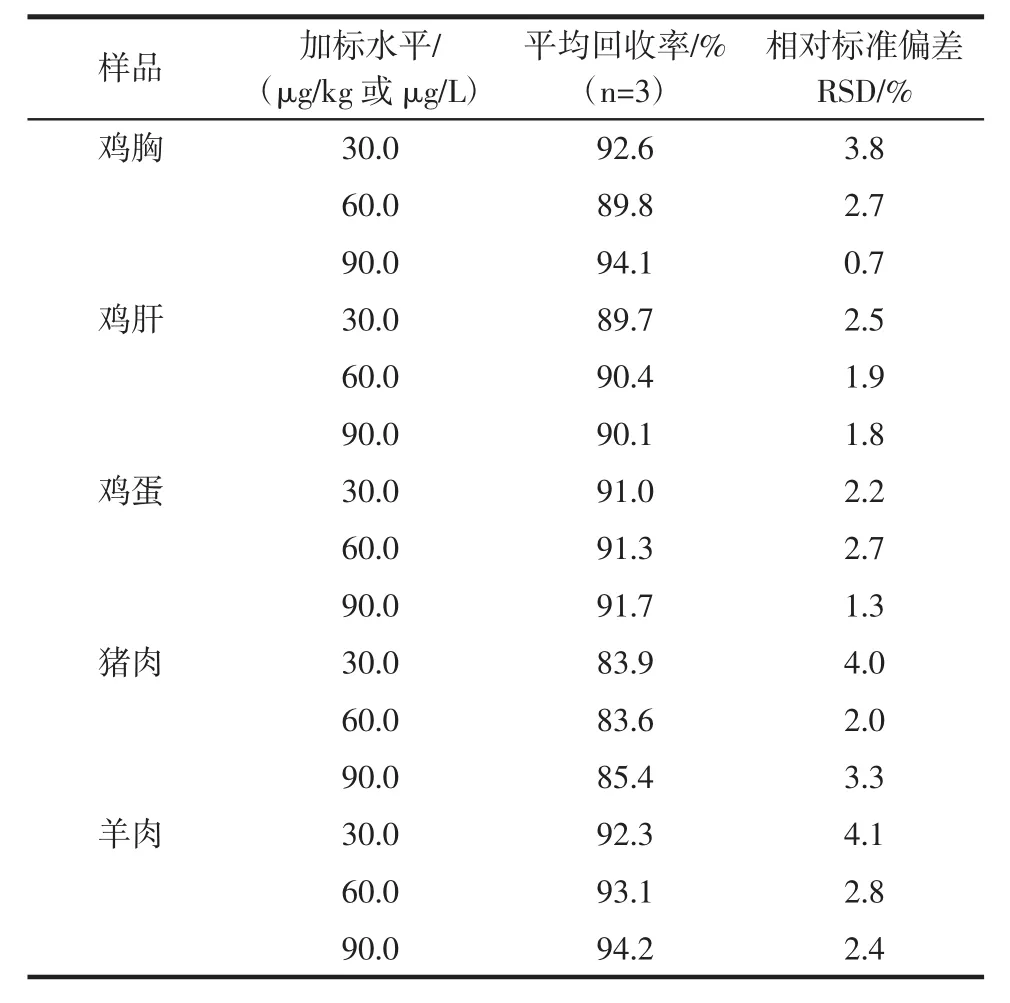
表3 樣品中金剛烷胺的回收率及相對標準偏差(n=3)Table 3 Recoveries and relative standard deviation(RSDs)of amantadine in different samples(n=3)
3 結論
本研究建立了一種固相萃取結合高效液相色譜串聯質譜法測定雞胸,雞肝,雞蛋,豬肉,羊肉中金剛烷胺殘留的方法。通過對各項參數的試驗,找出最佳的提取和凈化方法,對各項參數的調諧,使儀器檢測靈敏度達到最優化。對于分析物的線性范圍在0.1 μg/L~100.0 μg/L時的相關系數大于0.999;選定的5種動物源性食品中金剛烷胺的加標回收率在83.6%~94.2%之間;相對標準偏差小于4.0%。該方法具有簡便、快捷、精密度和準確性高的特點,可以作為檢驗檢疫行業動物源性食品中金剛烷胺殘留量的檢測方法。
[1] 葛孝忠,應黃慧,陳曉,等.金剛烷類藥物的研究進展[J].中國醫藥工業雜志,2003,34(11):583-586
[2] 包鴻俊.淺談抗病毒藥在獸醫上的應用[J].畜牧與獸醫,2002,34(6):1-3
[3] Chew CF,Guy A,Biggin PC.Distribution and Dynamics of Adamantanes in a Lipid Bilayer[J].Biophys J,2008,95:5627-5736
[4] Tominack R,Hayden F.Rimantadine hydrochloride and amantadine hydrochloride use in influenza A virus infections[J].Infectious disease clinics of North America,1987,1:459-478
[5] Wu YL,Chen RX,Xue Y,et al.Simultaneous determination of amantadine,rimantadine and memantine in chicken muscle using multi-walled carbon nanotubes as a reversed-dispersive solid phase extraction sorbent[J].Journal of chromatography B,Analytical technologies in the biomedical and life sciences,2014,965:197-205
[6] He G,Qiao J,Dong C,et al.Amantadine-resistance among H5N1 avian influenza viruses isolated in Northern China[J].Antiviral research,2008,77:72-76
[7] 李秋,王珊.抗病毒藥物的研究進展[J].醫藥導報,2011,6(30):732
[8]FDA Prohibits.Use of Antiviral Drugs in Poultry to Help Keep Drugs Effective for Humans[EB/OL].2009-06-l8.http://www.fda.Gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucml08620
[9] Farajzadeh MA,Nouri N,Alizadeh Nabil AA.Determination of amantadine in biological fluids using simultaneous derivatization and dispersive liquid-liquid microextraction followed by gas chromatography-flame ionization detection[J].Journal of chromatography B,Analytical technologies in the biomedical and life sciences,2013,940:142-149
[10]Leis HJ,Windischhofer W.Determination of memantine in human plasma by GC using negative ion chemical ionization MS detection after derivatization with a new reagent[J].Microchimica Acta,2012,178:309-314
[11]徐文彤.氣相色譜法測定硫酸金剛烷胺的含量[J].中國藥師,2007,10(1):1082-1083
[12]Van Der Horst FAL,Teeuwsen J,Holthuis JJM,et al.High-performance liquid chromatographic determination of amantadine in urine after micelle-mediated pre-column derivatization with 1-fluoro-2,4-dinitrobenzene[J].Journal of Pharmaceutical and Biomedical Analysis,1990,8:799-804
[13]Duh TH,Wu HL,Pan CW,et al.Fluorimetric liquid chromatographic analysis of amantadine in urine and pharmaceutical formulation[J].Journal of Chromatography A,2005,1088:175-181
[14]朱勝平,鐘華,何飛,等.高效液相色譜法測定鹽酸美金剛烷胺片的含量[J].中南藥學,2005,3(4):209-210
[15]裴璐,張愛兵.高效液相色譜-蒸發光散射檢測器法測定鹽酸金剛烷胺片中鹽酸金剛烷胺含量[J].中國藥業,2012,21(24):59-60
[16]Arndt T,Guessregena B,Hohla A,et al.Determination of serum amantadine by liquid chromatography-tandemmass spectrometry[J].Clinica Chimica Acta,2005,359:125-131
[17]陳慧華,韋敏玨,周煒,等.液相色譜-串聯質譜法測定動物組織中金剛烷胺和金剛乙胺的殘留量[J].質譜學報,2013,34(4):226-232
[18]劉正才,楊方,余孔捷.液相色譜-電噴霧串聯質譜法同時檢測雞組織中5種抗病毒類藥物的殘留量[J].色譜,2012,30(12):1253
[19]Yan H,Liu X,Cui F,et al.Determination of amantadine and rimantadine in chicken muscle by QuEChERS pretreatment method and UHPLC coupled with LTQ Orbitrap mass spectrometry[J].Journal of chromatography B,Analytical technologies in the biomedical and life sciences,2013,938:8-13
[20]Dou Y,Sun Y,Ren Y,et al.Simultaneous non-destructive determination of two components of combined paracetamol and amantadine hydrochloride in tablets and powder by NIR spectroscopy and artificial neural networks[J].J Pharm Biomed Anal 2005,37:543-549
[21]A Darwish,A S Khedr,H F Askal,et al.Simple and sensitive spectrophotometric methods for determination of amantaadine hydrochloride[J].Journal of Applied,2006,73(6):792-797
[22]Higashi Y,Nakamura S,Matsumura H,et al.Simultaneous liquid chromatographic assay of amantadine and its four related compounds in phosphate-buffered saline using 4-fluoro-7-nitro-2,1,3-benzoxadiazole as a fluorescent derivatization reagent[J].Biomedical Chromatography,2006,20:423-428
[23]Nour T Abdel-Ghani,Adel F Shoukry,Salwa H Hussein.Flow injection potentiometric determination of amantadine HCl[J].Journal of Pharmaceutical and Biomedical Analysis,2002,30(3):601-611
[24]Amorim CG,Araújo AN,Montenegro MCBSM,et al.Sequential Injection Lab-on-Valve Procedure for the Determination of Amantadine Using Potentiometric Methods[J].Electroanalysis,2007,19:2227-2233
Determination of Amantadine in Animal-derived Food by Combining SPE with HPLC-MS/MS
YUN Ya-guang1,2,CHI Yong-hong2,WANG Shuo1,*
(1.Tianjin University of Science and Technology,Tianjin 300457,China;2.Baotou Light Industry Vocational Technical College,Baotou 014035,Inner Mongolia,China)
An accurate and sensitive method for the detection of amantadine(AM)residues in animal-derived food products was developed by combining solid-phase extraction with high performance liquid chromatography-tandem mass spectrometry(SPE-HPLC-MS/MS).The Oasis MCX solid-phase extraction column was used for complex matrix purification and Agilent SB-C18 column(2.1 mm ×150 mm,3.5 μm)was employed for separation in liquid chromatography using a mobile phase consisting of 0.1%formic acid and acetonitrile(80∶20,volume ratio)with the flow rate 0.2 mL/min.The electrospray ionization (ESI)source in the positive mode and the multiple-reaction monitoring(MRM)mode were used for the quantitative analysis with external standard method.The results showed that the calibration curves were in good linearity for the AM ranged from 0.1 μg/L to 100 μg/L with the correlation coefficients(R2)> 0.999.The limits of detection(LODs)and the limits of quantification (LOQs)of this method were 1.0 μg/kg and 3.0 μg/kg respectively.The average recovery rates were from 83.6%to 94.2%with the good relative standard deviations (RSDs,RSD<4.0%,n=3)in chosen chicken mus cle,chicken liver,egg,beef and mutton samplesat three standard addition levelsof 30.0,60.0,and 90.0 μg/kg.
amantadine(AM);high performance liquid chromatography-tandem mass spectrometry(HPLCMS/MS);solid-phase extraction(SPE);animal-derived food;detection
10.3969/j.issn.1005-6521.2017.22.034
云雅光(1981—),女(蒙古),講師,碩士,研究方向:食品科學和食品檢測。
*通信作者:王碩(1969—),男,教授,博導,研究方向:食品科學。
2017-03-14
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
