有機硅改性環氧樹脂的光固化動力學與性能研究
2018-03-07 08:59:20胡芳友余周輝何西常趙培仲
裝備環境工程 2018年2期
胡芳友,余周輝,何西常,趙培仲
(1.海軍航空工程學院 青島校區 航空機械系,山東 青島 266041;2.陸軍工程大學,江蘇 徐州 221004)
作為一種高性能紫外光(UV)固化樹脂,脂環族環氧樹脂具有抗拉與抗壓強度高、耐高溫、耐紫外光老化及耐候性好等優點,但也存在質脆、耐熱性差、抗沖擊韌性差等缺點[1-3],限制了其更廣泛的應用,對脂環族環氧樹脂的增韌改性研究也成為今后的研究熱點。有機硅樹脂是以Si—O—Si鍵為主鏈、硅原子上連接有機基團的交聯型半無機高聚物,具有優異的柔韌性、耐候性、憎水性等特點,但也同樣存在粘接強度低、固化時間長、不便于大量應用等缺陷。通過共混或共聚的方式在環氧樹脂中添加適量比例的有機硅樹脂,在固化物中引入柔性Si—O鍵,既能降低環氧樹脂的內應力,改善環氧樹脂韌性、熱穩定性和耐濕熱性等,又能保持環氧樹脂自身的特性[4-5],從而使得有機硅改性環氧樹脂兼具有機硅樹脂和環氧樹脂的綜合性能,具有良好的發展前景。
目前有機硅改性環氧樹脂主要是利用有機硅化合物與熱固性環氧樹脂采用熱固化的方式完成的,采用光固化的方式實現有機硅改性環氧樹脂固化的報道還不多。這主要是由于紫外光的穿透能力有限造成的,對紫外光穿透不到的地方很難實現有效固化,大厚度樹脂的固化也成為紫外光固化的難點[6-10]。文獻[8-10]等研究結果指出,利用光引發前線聚合(PFP)是實現環氧樹脂大厚度快速固化的有效途徑。該技術利用光引發劑吸收紫外光分解引發聚合反應放熱,形成聚合前線并持續維持聚合反應直至體系全部固化。文中采用光引發劑與熱引發劑相結合的方式,選用端基或側鏈上帶有環氧基團有機硅樹脂(ES)與脂環族環氧樹脂(CEP)共混交聯。從理論上講,ES其自身就可與環氧樹脂實現開環聚合實現共聚反應,既可以較好地解決相容性問題,又可以與高性能紫外光樹脂實現光固化[11]。通過介電分析(dielectric analysis DEA)研究陽離子引發劑用量、熱引發劑用量對脂環族環氧樹脂/有機硅樹脂光固化動力學行為的影響,并利用熱失重分析(TG)、差示掃描熱(DSC)對固化物的熱性能進行分析,為紫外光固化環氧樹脂更廣泛的應用奠定基礎。
1 實驗
1.1 材料
實驗材料包括:脂環族環氧樹脂 UVR6128,雙((3,4-環氧環己基)甲基)己二酸脂,簡稱CEP,江蘇泰州泰特爾化工有限公司;有機硅樹脂ES-06,簡稱ES,吳江市合力樹脂有限公司;陽離子光引發劑820,二甲苯基碘鎓六氟磷酸鹽,姜堰市嘉晟科技有限公司;引發劑T,深圳初創新材料有限公司。該研究所用樹脂主要性能見表1。

表1 樹脂的主要性能
1.2 方案設計
設計了11種不同的紫外光固化樹脂配比試樣,實驗參數包括樹脂組分、輻照時間、引發劑含量與種類。為獲得各因素對光固化行為的影響,實驗設計方案見表2。
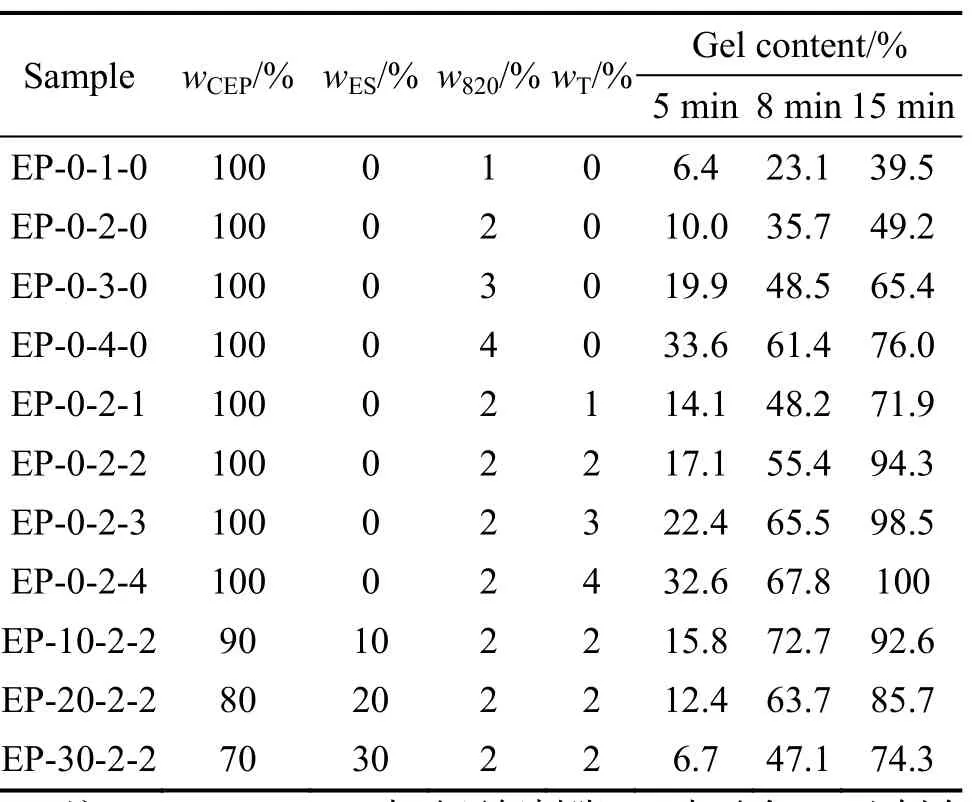
表2 實驗設計方案及測試結果
1.3 性能測試
1)采用NETZSHCH公司DEA28800A02型裝置測試樹脂固化反應過程。將不同配比樹脂涂敷于傳感器表面,涂層厚度控制為1 mm,然后置于1000 W高壓汞燈15 mm處。采樣頻率為20 Hz,樹脂起始溫度控制為25 ℃,光照一定的時間固化,測試樹脂介電性能隨時間的變化曲線。
2)采用上海鉅晶精密儀器制造有限公司的HV-1000IS型顯微硬度計測試樹脂固化后的表面硬度。測試前需將待測樹脂制成反光磨片試樣以便觀察,實驗加載砝碼質量為200 gf,通過測量四棱錐型的金剛石加壓試樣表面產生凹坑的對角線長度,計算待測物的纖維硬度值HV。
3)采用NETZSCH STA 449C熱分析儀測試樹脂體系的熱性能,包括熱失重(TG)和玻璃化轉變溫度(DSC),氮氣氣氛,升溫速率為10 ℃/min,測試溫度區間為 20~500 ℃。
2 結果與討論
2.1 DEA的光固化曲線分析
介電分析法(dielectric analysis, DEA)是通過感應信號振幅與相位的變化得出偶極子極化取向和離子移動的信息,測定樣品固化過程由于離子電導率的變化引起的介電損耗變化,從而得到高分子材料的離子黏度、固化速率、損耗因子等性質隨時間、溫度及頻率等微介電參數變化而變化的信息,實現固化過程的實時精確監測[12-13]。圖1為試樣EP-0-2-0損耗因子與離子黏度隨光照時間的變化曲線。從圖1可以看出,固化過程可分為三個階段,第一階段為0~2 min,此時離子黏度隨光照時間增加較為緩慢,屬于引發階段;第二階段為2~10 min,此時離子黏度快速上升,固化速率較快,屬于快速固化階段;超過10 min后,離子黏度與損耗因子隨光照時間無明顯變化,表明固化已基本結束。
離子黏度、介電損耗因子作為樹脂離子活動能力的表征,可間接表征樹脂的固化程度[14-15],該實驗選擇離子黏度來表征樹脂固化程度。為更好地分析各因素對固化過程的影響,對表2數據進行了歸一化處理。根據文獻[15],固化程度α可以表示為時間的函數:
式中:η∞為該實驗中所有樹脂體系最大離子黏度;tη為時間t時刻樹脂離子黏度;0η為樹脂初始離子黏度。根據式(1)計算所得實驗結果見表2。
2.2 引發劑種類與含量對動力學的影響
2.2.1 光引發劑
文中樹脂的起始黏度幾乎一致,初始溫度、光照強度及光照時間等因素均相同,因此僅有固化程度對離子黏度變化起主要作用。圖2為光引發劑820含量不同時各樹脂體系固化動力學曲線。從圖 2a可以看出,不同含量引發劑820的樹脂體系固化曲線外形大致相同,不同之處主要是引發固化反應所需的時間和快速固化階段固化速率。圖2b為引發前期樹脂固化動力學曲線,可以看出,當引發劑 820質量分數為1%時,引發固化反應大概需要3 min;當增加至4%時,引發固化反應只需1 min。可見,隨著引發劑820含量的增加,引發固化反應所需的時間逐漸縮短。當不同含量820樹脂體系進入快速固化階段,樹枝離子黏度隨固化時間快速增加,即反應劇烈程度逐漸增加。引發劑820濃度越大,離子黏度隨固化時間上升越快,且持續的時間略有增加。表現為相同光照時間下,引發劑820濃度越大,離子黏度越大,且當低濃度引發劑820的樹脂體系黏度趨于平緩時,高濃度引發劑樹脂體系離子黏度仍持續增加。當不同體系樹脂離子黏度不再變化時,表明固化已經結束。結合表2實驗結果可以得出:隨著光引發劑820含量的增加,引發固化反應所需時間逐漸縮短,快速固化階段固化速率逐漸增加,整體固化持續時間也有所增加,樹脂最終離子黏度也逐漸增加。因此,增加光引發劑820的含量對提高樹脂的固化效率是有益的。
2.2.2 熱引發劑
光引發前線聚合(PFP)理論認為只要形成聚合前線,即使停止光照,后續固化依舊可以依靠自身反應放熱促使熱引發劑的分解,維持單體的持續聚合直至體系全部固化[16]。該實驗中,不同樹脂體系在紫外光照射下,光引發劑820吸收光能分解產生超強酸引發環氧樹脂開環聚合。陽離子熱引發劑T本身并不吸收光能,但環氧樹脂的聚合屬于放熱反應,反應放熱與紫外光照射引起的樹脂溫度上升可促使引發劑 T的分解,引發劑T的分解進一步促進環氧樹脂的固化,固化反應放熱又可以促進引發劑T的分解,使得聚合反應持續進行。該實驗中的陽離子熱引發T就起到持續聚合的作用。圖3為樹脂體系中含2%質量分數820但引發劑T含量不同時的樹脂固化動力學曲線。從圖3a可以看出,隨著 T含量的增加,固化速率逐漸上升,最終離子黏度明顯增加,但增加幅度逐漸減緩。從圖3b可以看出,不同含量T的樹脂體系引發固化反應時間大致均為2 min,此時樹脂離子黏度隨光照時間增長均較為緩慢,表明T的加入對該階段影響較小。這主要是由于引發劑T本身不能吸收光能,而樹脂初始溫度較低未能促使引發劑T的分解,該階段主要由光引發劑820分解起聚合作用。當試樣進入快速固化階段后,與未添加T的試樣EP-0-2-0相比,添加引發劑 T的試樣在快速固化階段固化速率明顯增加,且固化速率隨T含量增加而增加,表現為相同光照時間下,樹脂離子黏度隨T含量增加而增加。當光照10 min后,試樣EP-0-2-0的離子黏度隨光照時間不再增加,說明固化過程已經結束。而含引發劑T的試樣在光照10 min后離子黏度仍逐漸增加,表明試樣固化過程仍在繼續,且隨著引發劑T含量的增加,這一作用效果更加明顯。特別是當T的質量分數增加至4%時,光照12 min后,樹脂離子黏度也基本不再增加,但此時樹脂離子黏度比其他試樣光照 15 min更大。結合表2數據對比分析僅含引發劑820的樹脂體系,由于紫外光穿透能力有限,對深層樹脂固化非常有限。即使當820的質量分數為4%時,光照15 min,樹脂最終固化程度也僅為76.0%。而當在含2%質量分數引發劑 820樹脂中加入 2%引發劑 T,光照 15 min,樹脂最終固化程度提高到 94.3%。盡管光引發劑820含量降低了,但最終固化程度得到明顯提高,表明引發劑T對后期深層樹脂起到很好的固化作用。從實驗結果可以得出:引發劑T的加入既能有效提高樹脂的固化程度,又能縮短固化時間,起到提高固化效率的作用。
2.3 樹脂組分
圖4為有機硅樹脂含量對樹脂固化程度的影響。從圖4可以看出,隨著有機硅樹脂含量的增加,固化速率與最終離子黏度逐漸下降,即樹脂固化程度逐漸下降。固化程度的下降可能有以下兩個原因:一方面有機硅含量的增加使得環氧樹脂濃度逐漸下降,參與開環聚合的環氧官能團活性中心減少(CEP環氧值為0.45~0.53 mol/100g,ES 環氧值為 0.03~0.08 mol/100g),固化速率下降,聚合反應放熱逐漸減少。又由前文分析可知,深層樹脂的固化主要依靠引發劑T的分解,反應放熱減少會降低引發劑T的引發效率,降低樹脂的固化效率。另一方面可能是由于有機硅含量的增加,兩者的固化速率差異性越來越大,相容性變差。過量的有機硅樹脂與環氧樹脂可能形成兩相結構,使得樹脂中活性基團參與反應的機會較少,降低整體固化速率。最終表現出引發聚合反應時間隨有機硅樹脂含量增加而逐漸增加,固化程度隨有機硅含量增加而逐漸下降。
2.4 固化物的硬度
紫外光照射15 min時,固化物維氏硬度隨有機硅含量的變化曲線如圖5所示。可以看出,隨著有機硅樹脂含量的增加,固化物的硬度逐漸下降,且下降速率隨ES含量增加而逐漸加快。當加入10%左右有機硅樹脂時硬度下降較為緩慢,超過此值,固化物硬度下降幅度快速增加。分析原因可能與2.3節分析ES含量的增加引起固化速率下降、相容性變差等因素均有一定的關系。
2.5 固化物的熱性能
表 3列出了不同樹脂配比固化物玻璃化轉變溫度。可以看出,隨著ES含量的增加,樹脂玻璃化轉變溫度逐漸下降,且下降幅度逐漸增大。原因可能有以下兩個方面:一方面ES含有柔順性較好的硅氧烷鍵段,玻璃化溫度較低,隨著ES含量的增加,聚合物更多地體現ES的特點;另一方面由2.3節分析可知,聚合物固化速率隨ES含量增加而逐漸下降,相同輻照時間下固化程度下降使得固化物固化程度下降。特別是當加入30%的ES后,有機硅樹脂與環氧樹脂相容性變差,固化效率變差。光照15 min樹脂固化程度僅為74.3%,較低的固化程度使得樹脂的耐熱性能下降明顯。總體而言,加入10%ES的共混樹脂玻璃化轉變溫度下降較小,表明此時ES/CEP樹脂達到較好的相容性,大分子之間形成互穿纏結降低了樹脂玻璃化轉變溫度的下降幅度。
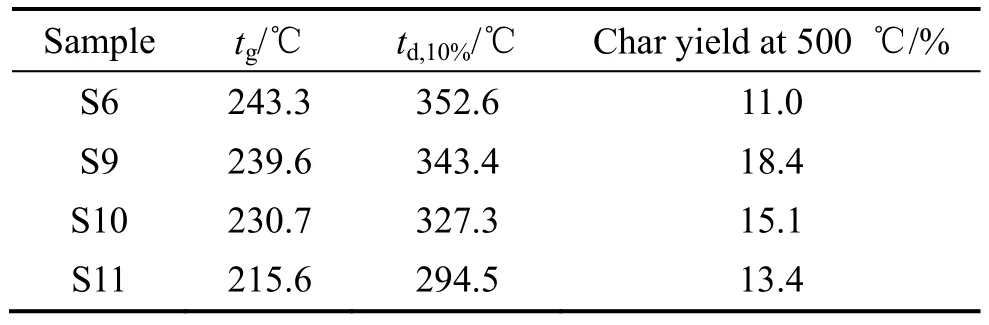
表3 ES/CEP樹脂的熱性能
圖6為不同配比樹脂體系熱失重曲線。從圖6可以看出,不同樹脂固化物熱失重過程可以分為兩個階段。第一階段主要在200~350 ℃之間,失重約為5%~15%,主要是引發劑和未固化的單體小分子造成的。表 3中td,10%表示固化物失重 10%時所對應的溫度,將此溫度定為初始分解溫度。從表3可以看出,樹脂初始分解溫度隨ES含量增加而逐漸降低,表明在低溫階段,樹脂的耐熱性能隨ES含量增加而逐漸下降,這主要是樹脂中未固化單體小分子含量隨ES含量增加而增加造成的。第二階段主要在350~450 ℃,該階段為快速失重區,主要是分子鍵的斷裂,大分子變成小分子。可以看出,高溫階段降解速率隨ES含量增加呈先下降后上升的趨勢,說明引入適量比例的 ES有利于提高固化物的耐高溫性能,這與加入適量比例有機硅樹脂固化物高溫殘炭率的提高結果是一致的。分析原因可能有以下兩個:一方面由于ES中的硅氧烷基團與環氧基團反應形成 Si—O—Si和 Si—O—C鍵,成為聚合物交聯網絡的一部分,Si—O—Si和Si—O—C鍵能遠大于 C—C鍵,使得聚合物材料的熱分解需要更多的能量。同時文獻[3,4]指出,Si—O—Si和Si—O—C分解后形成的SiO2會在材料表面形成一層致密的含硅保護層,有助于提高材料的熱穩定性。另一方面,ES含量超過一定值后,固化程度的下降也是固化物耐熱性能下降的重要原因。因此,有機硅樹脂含量增加后,如何提高預聚物的光固化效率還需要更多的研究。
3 結論
文中通過DEA分析了引發劑種類和有機硅樹脂含量對紫外光固化環氧樹脂動力學的影響,實現了有機硅改性環氧樹脂的有效固化。研究結果表明:
1)光引發劑820和熱引發劑T可以起到協同固化樹脂的作用。增加820的含量可以提高表層樹脂的固化速率,縮短引發時間。增加熱引發劑T可以對紫外線穿透不到的深層樹脂起很好的固化作用。光照15 min,試樣EP-0-2-2的固化程度可達94.3%,固化效果較為理想。
2)有機硅樹脂的加入降低了樹脂的光固化效率,固化物的玻璃化轉變溫度和低溫區域熱穩定性也有所下降,但高溫區域熱降解速率下降。總體而言,當加入10%左右有機硅樹脂時,固化物表現出較好的耐熱性能,500 ℃的殘炭率提高 67.3%,維氏硬度達31.75HV。
[1] 黎朝, 祈元春, 張彥慶, 等. 紫外光固化脂環族環氧丙烯酸酯涂料的制備及性能[J]. 功能高分子學報, 2012,1(3): 62-67.
[2] BULUT U, CRIVELLO J V. Investigation of the Reactivity of Epoxide Monomers in Photoinitiated Cationtic Polymerization[J]. Macromolecules, 2005, 38: 3584-3595.
[3] 王忠剛, 劉萬全, 趙林妮, 等.高性能脂環族環氧樹脂分子設計與合成研究進展[J]. 高分子通報, 2011, 25(9):13-21.
[4] 石新秀, 袁崇凱, 王傳萍, 等. 有機硅改性環氧樹脂的新進展[J]. 高分子通報, 2015, 25(4): 18-36.
[5] 魏振杰, 劉偉區, 李宏靜. 含磷有機硅雜化環氧樹脂固化體系性能研究[J]. 高分子學報, 2012, 2(2): 148-153.
[6] 劉紅波, 林峰, 肖望東, 等. 系列有機硅改性紫外光-熱混雜固化樹脂的表面性能[J]. 高分子材料科學與工程,2013, 29(4): 101-103.
[7] 常金, 李茂源, 洪義強, 等. 甲基苯基有機硅低聚體改性環氧樹脂的韌性及熱殘重[J]. 高分子材料科學與工程, 2013, 29(8): 72-75.
[8] 周建萍, 賈仕君, 傅萬里, 等. 紫外光引發環氧樹脂的下行前線聚合行為[J]. 高等學校化學學報, 2015, 36(5):1019-1024.
[9] CRIVELLO J. FALK V B, ZONCA M R. Photoactivated Frontal Polymerization of Oxetanes Using Optical Pyrometry[J]. Polymer, 2005, 46(26): 12109-12117.
[10] 崔艷艷, 楊建文, 曾兆華, 等. 前線聚合研究及應用進展[J]. 高分子通報, 2006(8): 44-51.
[11] 楊光, 黃鵬程. 氧原子對紫外光固化有機硅環氧樹脂的作用[J]. 宇航學報, 2008, 29(6): 2036-2040.
[12] SERNEK M, KAMKE F A. Application of Dielectric Analysis for Monitoring the Cure Process of Phenol Formaldehyde Adhesive[J]. Int J Adhes Adhes, 2007, 27(97):562-567.
[13] ZHOU Ya-wen, WEI Zhou, FU Han. Dielectric Analysis on Phase Transition and Micelle Shape of Polyoxyethylene Trisiloxane Surfactant in Dilute Aqueous Solution[J]. Chinese Chemical Letters, 2010, 10(22): 745-748.
[14] 尹海燕, 王成雙, 王玉婷, 等. 環氧瀝青的固化反應和微觀結構[J]. 高分子材料科學與工程, 2012, 28(11):30-33.
[15] 徐坤. 紫外光固化聚氨酯丙烯酸酯/ZrO2納米復合涂層的制備和性能研究[D]. 上海: 復旦大學, 2009.
[16] GUO D L, QING Z Y, CHANG C. Preparation of Porous Polyacrylamide Hydrogels by Frontal Polymerization[J].Polymer International, 2007, 56: 1016-1020.
