超高效液相色譜-串聯質譜法測定水產品中13種磺酰脲類除草劑殘留量
2018-03-13 07:52:32劉慧慧張華威董曉曉羅晶晶韓典峰薛敬林任傳博孫玉增
分析化學 2018年3期
關鍵詞:除草劑
劉慧慧 張華威 魏 瀟 董曉曉 羅晶晶 韓典峰 薛敬林 任傳博 黃 會 孫玉增
(山東省海洋資源與環境研究院,山東省海洋生態修復重點實驗室,煙臺 264006)
1 引 言
磺酰脲類除草劑是20世紀后期開發的一類廣譜、高效、高選擇性、低毒的除草劑,是目前世界上應用最廣泛、用量最大的除草劑之一[1~3],廣泛用于防治水稻、麥、大豆、玉米、油菜田雜草及草坪等非耕地雜草。該類除草劑不易揮發,在環境中降解較慢,可在環境中長期殘留[3]。磺酰脲類除草劑水溶性較強,在施用過程中容易隨著水循環直接或間接進入水環境中,影響水生植物的生長,進而影響水生動物的成長或繁殖,并且會隨著食物鏈富集,最終危害人類健康[4,5]。目前已有的檢測方法主要是針對農作物等植物源性食品[6]、水質[7]、土壤[8,9]等,而動物源性食品[10],尤其是水產品中的磺酰脲類除草劑殘留檢測研究較少。我國農業部公告第2032號[11]規定,自2015年12月31日起禁止使用氯磺隆,自2017年7月1日起禁止使用胺苯磺隆和甲磺隆; 我國國家標準GB2763-2016[12]規定了多種磺酰脲類除草劑在食品中的最大殘留限量。日本已對動物源性食品中多種磺酰脲類除草劑做出限量規定,如氟磺隆在豬肉和其它肉類的最大殘留限量為 0.05 mg/kg,砜嘧磺隆和丙苯磺隆在肝臟、豬肉、蛋和奶中的最大殘留限量為0.004 mg/kg,芐嘧磺隆在甲殼綱動物中最大殘留限量為50 μg/kg[13]。美國于2010年制定了噻吩磺隆(紅花籽中噻吩磺隆最大殘留量為0.05 mg/kg)殘留限量的法規[14]。因此,建立水產品中磺酰脲類除草劑多殘留檢測方法具有重要意義。
目前,磺酰脲類除草劑殘留的檢測方法主要有高效液相色譜法(HPLC)[15,16]、液相色譜-串聯質譜法(LC-MS/MS)[10,11,17,18]、毛細管電泳法(CE)[19,20]、酶聯免疫法(ELISA)[21]、高效薄層色譜法(HPTLC)[22]等,測定的樣品多為作物、土壤、水質等。水產品相對于作物、水質及土壤具有高脂肪、高蛋白等特點,較畜禽類等其它動物源性食品而言,水產品的基質更為復雜,不同種類水產品基質差別較大,檢測方法需要滿足不同水產品基質的特性,以高效地提取目標物、去除雜質、減少干擾。本研究采用固相萃取法對樣品進行凈化、富集,超高效液相色譜-串聯質譜法對樣品溶液進行分離、檢測,建立了水產品中13種磺酰脲類除草劑的快速、靈敏、準確的檢測方法,以滿足水產品樣品中農藥殘留分析的要求。
2 實驗部分
2.1 儀器與試劑
UPLC-Quattro Premier超高效液相色譜-串聯質譜儀(美國Waters公司); 高速控溫離心機(德國Sigma公司); N-EVAPTM112氮吹儀(美國Qrganomation Associates公司); Milli-Q Gradient超純水儀(法國Millipore公司); KQ-600E超聲波清洗器(昆山市超聲儀器有限公司); 旋轉蒸發儀(德國IKA公司); AccuPrep MPS凝膠色譜系統(美國J2公司)。
甲醇、乙腈、二氯甲烷、環己烷(色譜純,德國Merck公司); 甲酸、乙酸(阿拉丁公司); MAX固相萃取柱、HLB固相萃取柱(3 mL/60 mg,美國Waters公司); C18固相萃取柱(3 mL/500 mg,美國Waters公司)。13種磺酰脲類除草劑(氟胺磺隆、氯吡嘧磺隆、吡嘧磺隆、氯嘧磺隆、胺苯磺隆、芐嘧磺隆、煙嘧磺隆、醚苯磺隆、苯磺隆、甲基噻吩磺隆、甲磺隆、甲嘧磺隆、氯磺隆)標準品(純度均≥98%,德國Dr. Ehrenstorfer公司); 實驗用水為超純水。
單標儲備溶液: 稱取適量磺酰脲類除草劑標準品,用甲醇溶解,配成100 mg/L標準儲備溶液,于4℃保存。混合標準溶液: 分別準確移取適量上述單標儲備溶液,以乙腈定容,配制成質量濃度為1 mg/L 的混合標準溶液。于4℃保存,臨用時稀釋成所需濃度。
2.2 樣品提取與凈化
2.2.1樣品處理鯉魚、南美白對蝦、中華絨鰲蟹、文蛤和海參購自農貿市場,分別取其可食部分進行勻質,4℃保存待用。
2.2.2提取方法準確稱取5 g(精確至0.01 g)樣品,各加入25 mL乙酸乙酯,渦旋混勻30 s,超聲30 min,4000 r/min離心10 min,上清液分別轉移至100 mL雞心瓶中, 殘渣用20 mL乙酸乙酯按照同樣步驟再提取一次, 合并上清液。于40℃旋轉蒸發至干,5 mL甲醇-水(1∶4,V/V)溶解殘渣,待用。
2.2.3凈化方法取MAX固相萃取小柱,依次用5 mL甲醇、5 mL水活化,加入上述復溶液,依次用5 mL甲醇-水(1∶4,V/V)、5 mL甲醇淋洗,棄去全部流出液,用5 mL乙酸-甲醇(1∶50,V/V)洗脫。40℃氮氣吹干,用含0.1%甲酸-乙腈混合溶液(6∶4,V/V)1 mL溶解殘渣,加3 mL正己烷,渦旋混勻30 s,4000 r/min離心10 min,濾液過0.22 μm有機濾膜,待測。
2.3 色譜條件
ACQUITYTMUPLC BEH C18色譜柱(100 mm×2.1 mm i.d.,1.7 μm); 流動相A為乙腈,B為0.1%甲酸; 流速: 0.25 mL/min; 進樣量: 10 μL; 柱溫: 30℃; 梯度洗脫程序: 0~2.00 min, 20% A; 2.00~7.00 min, 90% A; 7.00~10.0 min, 20% A。
2.4 質譜條件
電離方式: ESI+; 掃描方式: 多反應監測(MRM); 掃描時間: 0.1 ms; 電離電壓3.00 kV; 離子源溫度: 110℃; 錐孔反吹氣流量: 50 L/h; 脫溶劑氣溫度: 350℃; 脫溶劑氣: N2; 脫溶劑氣流量: 700 L/h。母離子、子離子、錐孔電壓、碰撞能量等質譜參數見表1。
表1 13種磺酰脲類除草劑的多反應監測(MRM)參數
Table 1 Parameters for 13 kinds of sulfonylurea herbicides in multi-reactions monitoring(MRM) mode
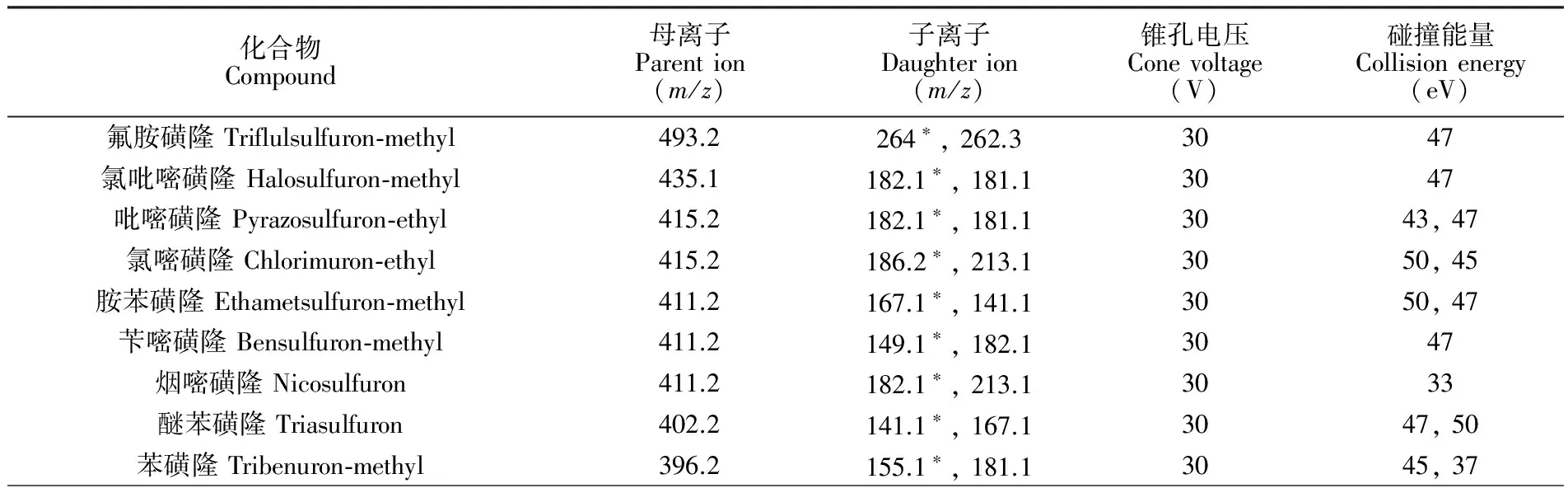
化合物Compound母離子Parention(m/z)子離子Daughterion(m/z)錐孔電壓Conevoltage(V)碰撞能量Collisionenergy(eV)氟胺磺隆Triflulsulfuron?methyl493.2氯吡嘧磺隆Halosulfuron?methyl435.1吡嘧磺隆Pyrazosulfuron?ethyl415.2氯嘧磺隆Chlorimuron?ethyl415.2胺苯磺隆Ethametsulfuron?methyl411.2芐嘧磺隆Bensulfuron?methyl411.2煙嘧磺隆Nicosulfuron411.2醚苯磺隆Triasulfuron402.2苯磺隆Tribenuron?methyl396.2264?,262.33047182.1?,181.13047182.1?,181.13043,47186.2?,213.13050,45167.1?,141.13050,47149.1?,182.13047182.1?,213.13033141.1?,167.13047,50155.1?,181.13045,37
續表1(Continued to Table 1)

化合物Compound母離子Parention(m/z)子離子Daughterion(m/z)錐孔電壓Conevoltage(V)碰撞能量Collisionenergy(eV)甲基噻吩磺隆Thifensulfuron?methyl388.1甲磺隆Metsulfuron?Methyl382.2甲嘧磺隆Sulfometuron?Methyl364.9氯磺隆Chlorsulfuron358.2167.1?,205.13040,47167.2?,199.23040,43150.1?,199.13045,43141.1?,167.13050,43注:?代表定量離子。Note:?representquantitativeion.
3 結果與討論
3.1 UFLC-MS/MS 分析條件的優化
3.1.1流動相的確定磺酰脲類除草劑呈酸性,在流動相中加入適量甲酸,可以提高霧化效率,改善峰形。本研究所用的流動相中的有機相分別采用乙腈和甲醇,水相分別采用純水、0.1%甲酸及5 mmol/L乙酸銨-0.1%甲酸溶液,對比了13種目標化合物和干擾物在不同組分流動相中的分離特性,發現采用乙腈和0.1%甲酸梯度洗脫時,13種目標物響應值及峰形最佳。鯉魚空白加標樣品中13種磺酰脲除草劑(10.0 μg/kg)的二級質譜總離子流色譜圖見圖1。
3.1.2特征離子對的選取采用正離子掃描模式,優化碰撞能量,采用子離子掃描模式,選擇響應最高的兩個子離子作為特征子離子。以甲嘧磺隆為例,其易形成m/z364.9的[M+1]+離子,碰撞后響應最高的子離子為m/z150.1和m/z199.1,作為特征子離子。

圖1 鯉魚空白樣品(KB)和加標(10.0 μg/kg)樣品(TJ)的13種磺酰脲類除草劑的總離子流圖Fig.1 Total ion chromatograms of 13 kinds of sulfonylurea herbicides in blank (KB) and spiked (10.0 μg/kg) (TJ) carp samples
3.2 樣品前處理條件的優化
3.2.1提取條件的確定根據磺酰脲類除草劑的特點,本研究采用加標回收法對提取溶劑進行優化。在鯉魚空白樣品中加入13種磺酰脲類除草劑混合標準溶液,加標水平均為10.0 μg/kg,分別以乙酸乙酯、乙腈、甲醇和丙酮4種有機溶劑為提取液,比較不同提取溶劑對13種磺酰脲類除草劑提取效率的影響,結果見表2。對13種目標物,乙酸乙酯與乙腈的提取效率均大于70%,優于甲醇及丙酮; 然而乙腈作為提取液時,所提取溶液中水含量高,提取溶液難以濃縮,并且乙腈提取液在旋轉蒸發濃縮時容易暴沸,不但增加了濃縮的難度也容易降低回收率。因此選擇乙酸乙酯作為提取溶劑。
表2 4種提取溶劑對鯉魚中13種磺酰脲類除草劑(10.0 μg/kg)的平均回收率(n=3)
Table 2 Recoveries of 13 kinds of sulfonylurea herbicides (10.0 μg/kg) in carp using 4 kinds of extraction solvents (n=3)

化合物Compound乙酸乙酯Ethylacetate(%)乙腈Acetonitrile(%)甲醇Methylalcohol(%)丙酮Acetone(%)氟胺磺隆Triflulsulfuron?methyl97.1±5.793.7±6.583.5±8.770.8±10.4氯吡嘧磺隆Halosulfuron?methyl99.5±5.296.2±9.464.9±6.350.2±7.1吡嘧磺隆Pyrazosulfuron?ethyl89.7±9.495.9±6.561.0±8.884.7±6.4氯嘧磺隆Chlorimuron?ethyl75.1±4.674.6±5.377.3±7.569.5±7.6胺苯磺隆Ethametsulfuron?methyl102±8.494.0±8.360.1±8.355.0±6.9芐嘧磺隆Bensulfuron?methyl86.0±7.594.2±7.262.8±8.483.5±8.8煙嘧磺隆Nicosulfuron102±4.276.7±5.874.6±6.959.4±7.5醚苯磺隆Triasulfuron94.6±5.295.5±9.181.1±3.769.2±9.2苯磺隆Tribenuron?methyl94.1±5.779.8±8.978.0±6.958.9±3.0甲基噻吩磺隆Thifensulfuronmethyl80.9±4.394.6±2.869.3±9.683.3±8.5甲磺隆Metsulfuron?Methyl79.5±5.473.0±2.478.1±9.775.7±4.6甲嘧磺隆Sulfometuron?Methyl79.3±5.394.7±6.258.2±7.774.6±8.7氯磺隆Chlorsulfuron94.8±8.388.2±7.281.8±6.548.0±3.6
3.2.2凈化方法的選擇及優化常用的前處理凈化方法有固相萃取法、GPC凝膠色譜法、液-液凈化法及基質固相分散法等。由于水產品樣品基質中蛋白質和脂類含量較為豐富,干擾組分多,對凈化方法要求較高,液-液凈化法及基質固相分散法難以達到凈化要求,其凈化液仍需進一步凈化方可用于儀器檢測。因此本研究通過實驗考察對比了MAX固相萃取柱、C18固相萃取柱、HLB固相萃取柱和GPC凝膠色譜對草魚、中華絨鰲蟹樣品提取液的凈化效果。實驗結果表明,使用MAX及HLB固相萃取柱凈化樣品時,回收率均大于70%,且兩種小柱的凈化效果相當,但具有親水親脂平衡反向吸附特性的HLB小柱,在中華絨鰲蟹等脂類含量高的樣品過柱時,提取溶液的流出速度明顯小于MAX小柱,受提取液中樣品基質影響較明顯; 并且磺酰脲類除草劑呈酸性,采用混合型陰離子交換反相吸附劑的MAX小柱對于酸性化合物具有更高的選擇性和靈敏度; C18固相萃取柱的凈化效果與MAX及HLB接近,但甲基噻吩磺隆的回收率僅為53.2%,遠低于MAX及HLB固相萃取柱; GPC凝膠色譜雖然在去除大分子蛋白質及脂類方面具有明顯優勢,但由于磺酰脲類除草劑分子量也較大,導致凈化液同流出物中仍有較多大分子干擾,凈化效果較差。綜合考慮以上結果,本研究選擇MAX固相萃取柱作為凈化柱。
根據磺酰脲類除草劑極性較強的特征,本研究分別選用乙酸-甲醇(1∶50,V/V)、乙腈、正己烷-丙酮(1∶1,V/V)作為MAX固相萃取柱的洗脫溶劑。實驗表明,5 mL乙酸-甲醇(1∶50,V/V)即可使13種磺酰脲類除草劑的回收率達到70%以上,洗脫效果最好,洗脫速度及回收率均明顯高于乙腈和正己烷-丙酮(1∶1,V/V)。因此,本研究選擇乙酸-甲醇混合溶液(1∶50,V/V)為MAX固相萃取柱的洗脫溶劑。
3.3 基質效應
本研究對基質效應[23]進行了評價,采用空白鯉魚及中華絨鰲蟹樣品的提取溶液作為基質,分別配制13種磺酰脲類除草劑的系列濃度基質標準溶液,同時使用流動相配制相應的系列濃度標準溶液。采用對比基質標準曲線斜率與溶劑標準曲線斜率比值的方法評價基質效應[23,24]。結果表明,13種磺酰脲類除草劑基質效應范圍在41.1%~88.3%之間,均存在基質抑制效應。其中,氟胺磺隆、氯吡嘧磺隆、吡嘧磺隆、氯嘧磺隆、甲基噻吩磺隆、甲磺隆、氯磺隆7種化合物存在明顯的基質抑制效應。為消除或減弱基質效應干擾,本研究采用相應空白基質溶液配制標準溶液繪制校正曲線。
3.4 線性關系和靈敏度
取適量磺酰脲類除草劑混合標準溶液,分別用鯉魚、南美白對蝦、中華絨鰲蟹、文蛤和海參空白樣品基質溶液配制成5.0、10.0、20.0、50.0 和100.0 μg/L系列濃度梯度的標準溶液,按照2.3節及2.4節的條件進行測定,以各組分響應值為縱坐標,質量濃度為橫坐標,進行線性回歸分析。結果表明,13種磺酰脲類除草劑線性良好,結果見表3。采用標準溶液加入法,在鯉魚、南美白對蝦、中華絨鰲蟹、文蛤和海參空白樣品中添加一定濃度的標準溶液進行測定,以定量離子信噪比S/N=3對應的濃度為樣品的檢出限(LOD),S/N=10為標準確定定量限(LOQ),得到13種除草劑的LOD為1.0 μg/kg,LOQ為2.0 μg/kg。
表3 13種磺酰脲類除草劑的線性方程、線性范圍和相關系數
Table 3 Linear equations, linearity range and correlation coefficients of 13 kinds of sulfonylurea herbicides
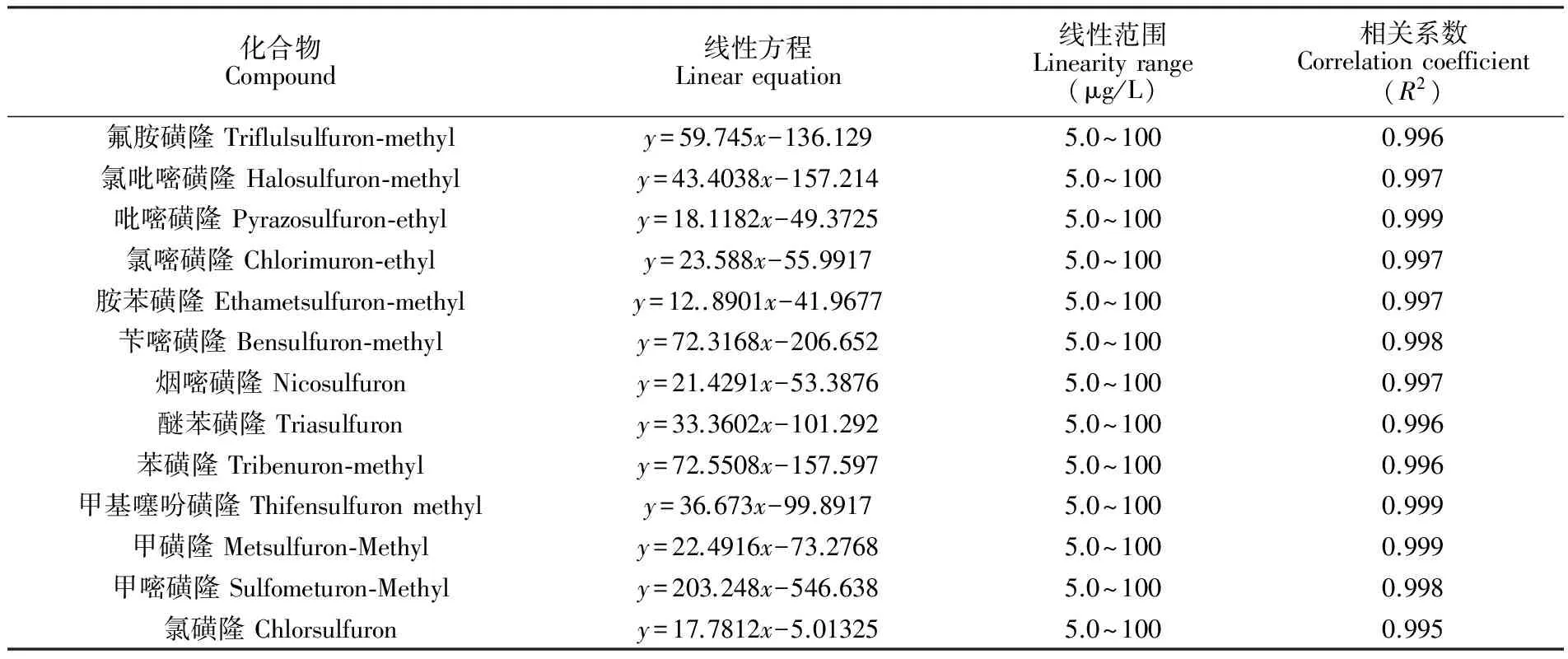
化合物Compound線性方程Linearequation線性范圍Linearityrange(μg/L)相關系數Correlationcoefficient(R2)氟胺磺隆Triflulsulfuron?methyly=59.745x-136.1295.0~1000.996氯吡嘧磺隆Halosulfuron?methyly=43.4038x-157.2145.0~1000.997吡嘧磺隆Pyrazosulfuron?ethyly=18.1182x-49.37255.0~1000.999氯嘧磺隆Chlorimuron?ethyly=23.588x-55.99175.0~1000.997胺苯磺隆Ethametsulfuron?methyly=12..8901x-41.96775.0~1000.997芐嘧磺隆Bensulfuron?methyly=72.3168x-206.6525.0~1000.998煙嘧磺隆Nicosulfurony=21.4291x-53.38765.0~1000.997醚苯磺隆Triasulfurony=33.3602x-101.2925.0~1000.996苯磺隆Tribenuron?methyly=72.5508x-157.5975.0~1000.996甲基噻吩磺隆Thifensulfuronmethyly=36.673x-99.89175.0~1000.999甲磺隆Metsulfuron?Methyly=22.4916x-73.27685.0~1000.999甲嘧磺隆Sulfometuron?Methyly=203.248x-546.6385.0~1000.998氯磺隆Chlorsulfurony=17.7812x-5.013255.0~1000.995
3.5 準確度和精密度
準確稱取5.0 g(精確至0.01 g)空白鯉魚、中華絨鰲蟹和文蛤肉糜,加入適量磺酰脲類除草劑混合標準溶液,充分混勻,加標濃度分別為2.0、10.0和20.0 μg/kg,按本方法進行測定,每個加標濃度進行6次平行測定。結果表明,13種磺酰脲類除草劑在鯉魚中的加標回收率在85.1%~105.7%之間,相對標準偏差(RSD)為2.1%~10.8%; 在中華絨鰲蟹中的加標回收率在75.4%~118.3%之間,RSD為5.6%~14.5%; 在文蛤中的加標回收率在83.2% ~ 110.5%之間,RSD為4.7%~12.4%。本方法的準確度與精密度能滿足水產品中污染物的分析要求。
3.6 實際樣品分析
使用本方法對本實驗室常規檢測留存的養殖草魚、鯉魚、海參、南美白對蝦、大菱鲆等5種10個樣品及市場購買的中華絨鰲蟹2個樣品進行測定,均未檢出13種磺酰脲類除草劑。
將體重為(100±25) g健康中華絨鰲蟹暴露于氯吡嘧磺隆濃度為1.0 mg/L的溶液中(室溫(20±1)℃),分別于暴露后第24、48和72 h對中華絨鰲蟹進行采樣,取其肌肉及性腺,使用本方法測定其中氯吡嘧磺隆含量。結果表明,暴露第24、48和72 h中華絨鰲蟹可食組織中氯吡嘧磺隆含量分別為6.20、 12.1和16.6 μg/kg。中華絨鰲蟹樣品中氯吡嘧磺隆的選擇離子流圖見圖2。
上述結果表明, 本方法能夠滿足我國以及日本、美國對該類除草劑的限量要求,方法高效、經濟、準確、靈敏,適用于水產品中13種磺酰脲類除草劑的定量及確證分析。
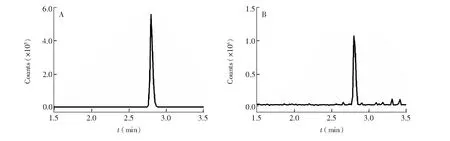
圖2 中華絨鰲蟹中氯吡嘧磺隆的定量離子圖(A)與定性離子圖(B)Fig.2 Quantitative ion chromatogram (A) and qualitative ion chromatogram (B) of halosulfuron-methyl in Eriocheir sinensis
1 Christopher J T, Burnet M, Powles S B, Holtum J A M, Liljegren D R. 1InternationalWeedControlCongress,1992, 12(3): 269-83
2 HU Kai-Feng, SUN Tao, LIU Sheng-Hong, QIAO Kun-Yun.JournalofInstrumentalAnalysis,2010, 29(11): 1216-1220
胡開峰,孫 濤,劉圣紅,喬坤云. 分析測試學報,2010, 29(11): 1216-1220
3 Wang B, Kong D, Lu J, Zhou Q.Environ.Sci.Pollut.Res.,2015, 22(5): 3847-3855
4 YANG Ren-Bin, LIU Yi-Hua, QIU Jian-Xia, ZHENG Li-Ying, PENG Juan-Ying.JournalofHunanAgriculturalUniversity(NaturalSciences),2007, 33(1): 96-100
楊仁斌,劉毅華,邱建霞,鄭麗英,彭娟瑩. 湖南農業大學學報(自然科學版),2007, 33(1): 96-100
5 Baker S E, Olsson A O, Needham L L, Barr D B.Anal.Bioanal.Chem.,2005, 383(6): 963-976
6 DING Fei, LI Fan-Zhu, CHU Xiao-Gang, ZHANG Feng, LING Yun, SUN Li, YANG Min-Li, LV Quan-Fu, XU Cheng-Bao.JournalofInstrumentalAnalysis,2011, 30(1): 53-57
丁 菲, 李范珠, 儲曉剛, 張 峰, 凌 云, 孫 利, 楊敏莉, 呂泉福, 許成保. 分析測試學報,2011, 30(1): 53-57
7 ZHAO Yong-Gang, ZHANG Xiang-Zhi, HU Guan-Jiu, ZHANG Yong, ZHOU Chun-Hong, LI Juan, ZHANG Bei-Bei.JournalofAnalyticalScience,2008, 24(3): 287-290
趙永剛, 張祥志, 胡冠九, 章 勇, 周春宏, 李 娟, 張蓓蓓. 分析科學學報,2008, 24(3): 287-290
8 YE Gui-Biao, ZHANG Wei, CUI Xin, PAN Can-Ping, JIANG Shu-Ren.ChineseJ.Anal.Chem.,2006, 34(9): 1207-1212
葉貴標, 張 微, 崔 昕, 潘燦平, 江樹人. 分析化學,2006, 34(9): 1207-1212
9 Bossi R, Vejrup K, Jacobsen C S.J.Chromatogr.A,1999, 855(2): 575-582
10 LIU Jin-Xia, ZHANG Ying, DING Li, LIU Xiao-Xia, HUANG Zhi-Qiang, CHEN Bo, WANG Li-Bing.ChineseJ.Anal.Chem.,2011, 39(5): 664-669
劉錦霞, 張 瑩, 丁 利, 劉曉霞, 黃志強, 陳 波, 王利兵. 分析化學,2011, 39(5): 664-669
11 Ministry of Agriculture of the People's Republic of China.AnnouncementNo. 2032ofMinistryofAgricultureofthePeople'sRepublicofChina.2013
中華人民共和國農業部. 中華人民共和國農業部公告 第2032號.2013
12 GB2763-2016.NationalFoodSafetyStandard-MaximumResidueLimitsforPesticidesinFood, National Standards of the People's Republic of China
食品安全國家標準-食品中農藥最大殘留限量. 中華人民共和國國家標準. GB2763-2016
13 Department of Food Safety, Ministry of Health, Labour and Welfare.TheJapanesePositiveListSystemforAgriculturalChemicalResiduesinFoods.2017
14 EPA-HQ-OPP-2009-0134.FederalRegister,2010, 75(71): 19272-19277
15 SUI Kai, LI Jun, WEI Feng, CHU Xiao-Gang, ZHAO Shou-Cheng, WANG Yu-Ping.ChineseJournalofChromatography,2006, 24(2): 152-156
隋 凱, 李 軍, 衛 鋒, 儲小剛, 趙守成, 王玉萍. 色譜,2006, 24(2): 152-156
16 Wu Q H, Wang C, Liu Z M, Wu C X, Zeng X, Wen J L, Wang Z.J.Chromatogr.A,2009, 1216: 5504-5510
17 QI Yan, LI Shu-Juan, ZHAN Chun-Rui, PENG Tao.ChineseJ.Anal.Chem.,2004, 32(11): 1436-1440
祁 彥, 李淑娟, 占春瑞, 彭 濤. 分析化學,2004, 32(11): 1436-1440
18 LI Na, LI Hui, SHAO Hui, LIU Lei, ZHANG Yu-Ting, GUO Yong-Ze.ChineseJournalofChromatography,2011, 29(4): 346-352
李 娜, 李 輝, 邵 輝, 劉 磊, 張玉婷, 郭永澤. 色譜,2011, 29(4): 346-352
19 Dinelli G, Vicari A, Brandolini V.J.Chromatogr.A,1995, 700(1): 201-207
20 Daniel D, dos Santos V B, Vidal D T, Lago C L.FoodChem.,2015, 175: 82
21 Piet S, Bossi R, Streibig J C.PestManagementSci.,2000, 56(7): 637-643
22 SHI Ya-Qin, YAO Jing, ZHANG Qi-Ming, JIN Shao-Hong.ChineseJournalofPharmaceuticalAnalysis,2007, (1): 36-39
施亞琴, 姚 靜, 張啟明, 金少鴻. 藥物分析雜志,2007, (1): 36-39
23 Kittlaus S, Schimanke J, Kempe G, Speer K.J.Chromatogr.A,2011, 1218(46): 8399-8410
猜你喜歡
世界農藥(2019年3期)2019-09-10 07:04:10
今日農業(2019年15期)2019-01-03 12:11:33
現代園藝(2017年19期)2018-01-19 02:50:21
長江蔬菜(2016年10期)2016-12-01 03:05:27
獸醫導刊(2016年12期)2016-05-17 03:51:29
現代農業(2016年5期)2016-02-28 18:42:36
雜草學報(2015年2期)2016-01-04 14:58:05
種業導刊(2016年9期)2016-01-03 01:27:14
營銷界(2015年23期)2015-02-28 22:06:18
營銷界(2015年22期)2015-02-28 22:05:11
