過渡金屬催化C(sp2)-H鍵活化構建手性碳原子中心的研究進展
2018-04-19 08:42:52熊儒恒
浙江化工 2018年3期
熊儒恒,帥 棋
(浙江工業大學長三角綠色制藥協同創新中心,浙江 杭州 310014)
0 前言
手性碳原子中心是自然界中普遍存在的化學結構,廣泛存在于天然產物[1]、醫藥中間體[2]等多種化合物中。因此,如何有效地構建手性碳原子中心,越來越受到有機合成研究人員的密切關注。
碳氫鍵(C-H)活化反應是當前有機反應方法學研究的一個重要領域。直接利用碳氫鍵活化反應構建手性碳原子中心是一種十分簡潔高效的方法。在過去的十幾年中,隨著對過渡金屬催化碳氫鍵活化反應的研究,C(sp2)-H鍵活化反應的研究不斷取得重大進展。然而,如何通過選取合適的催化體系實現不對稱C(sp2)-H鍵活化構建手性碳原子中心仍然是困擾研究人員的難題之一,也因此正得到越來越多的重視。鑒于目前關于此類反應的綜述并不能完全闡明該類反應的研究進展,本文在前人的工作基礎之上[3],通過整理自2013年以來的過渡金屬催化不對稱C(sp2)-H鍵活化構建手性碳原子中心的相關文獻,著重介紹了該類反應的最新研究進展,希望能使相關研究人員對該反應的機理和應用有更深的理解和認識。
1 過渡金屬催化C(SP2)-H鍵活化構建手性中心的反應
Hou等人[4]報道了一起采用手性鈧復合物催化C(sp2)-H鍵活化引入手性中心的對映選擇性反應(圖1)。該反應以2-位取代吡啶和端烯烴化合物為底物,成功構建手性叔碳手性中心,產物的ee值最高可達88%。

圖1
Gong課題組[5]報道了一起采用 Ru(II)或者Rh(II)復合物與喹啉衍生物為催化體系,以3-重氮氧化吲哚化合物,吲哚和硝基烯烴化合物為底物的三組分不對稱C(sp2)-H鍵活化官能團化反應(圖2)。該反應的反應效率高,ee值最高達97%,且產物為具有光學活性的3,3-二取代吲哚類化合物。

圖2
胡文浩課題組[6]報道了一起以手性磷酸化合物和Rh為催化體系,實現了對苯胺衍生物、重氮化合物和亞胺化合物的不對稱C(sp2)-H鍵活化反應(圖3-a)。該反應在 Rh 催化 C(sp2)-H 鍵活化時,產生非手性中間體,在其質解之前發生手性磷酸催化的不對稱Mannich反應,得到具有兩個連續手性中心的產物。該反應的不足之處在于,芳烴底物只適用于苯胺衍生物。之后,周其林課題組[7]報道了類似的反應,也完成了重氮化合物對苯胺衍生物的不對稱C(sp2)-H鍵活化官能團化反應(圖3-b)。這兩類反應產物的ee值均較高,最高可達99%。
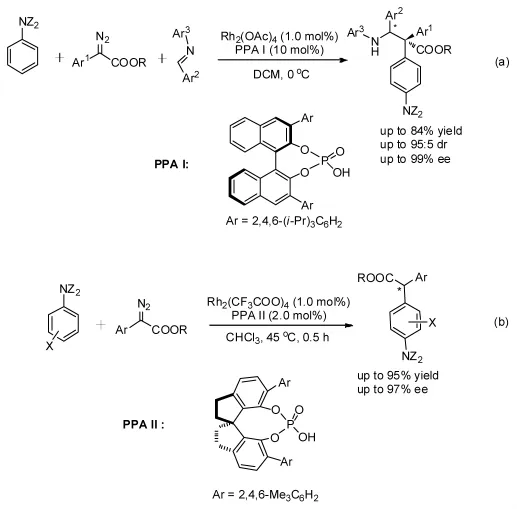
圖3
Yu課題組[8]設計了一系列手性氨基酸衍生物作為配體并用于Pd催化的具有對映選擇性的碳氫鍵活化反應當中。在反應中,Yu等人以環丁基甲酸衍生物和芳基硼試劑為反應物,經過不對稱碳氫鍵活化交叉偶聯反應構建季碳手性中心(圖4)。該反應產率高,對映選擇性高達92%。同時研究人員發現,環丁基甲酸衍生物上的吸電子酰胺取代基在偶聯反應中可充當導向基團,而且手性配體上大位阻和吸電子基團的結構對產物的對映選擇性十分重要,這為下一步研究該反應指明了方向。

圖4
Shibata等[9]在合成三環吲哚類化合物時,發現了一起Ir催化的對映選擇性C(sp2)-H鍵烷基化反應 (圖5)。該反應發生在吲哚化合物的C2位置,具有很高的對映選擇性。在該反應中,研究人員以陽離子Ir復合物和手性膦配體為催化體系合成了一系列具有手性中心的化合物。該反應產率高,反應產物的ee值最高可達98%。
圖5
Yoshikai課題組[10]報道了一起以苯乙烯化合物為底物,金屬Co催化的吲哚化合物C(sp2)-H鍵不對稱烷基化反應(圖6)。在該反應中,研究人員發現,采用亞磷酰胺化合物作為手性配體,反應效果最好。當苯乙烯底物帶有給電子取代基時,其反應產物的對映選擇性較缺電子類苯乙烯化合物高。
Yamamoto和合作者[11]在研究Togni課題組[12]早期報道的降冰片烯的不對稱C(sp2)-H鍵烷基化工作時發現,采用(R,R)-S-MeBIPAM作為雙齒配體進行降冰片烯烷基化反應,產物ee值最高可達99%(圖7),且酮化合物和酰胺類化合物均適用于本反應。
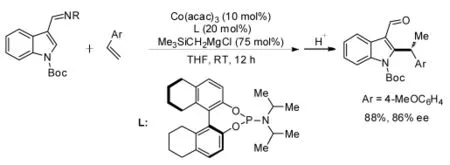
圖6

圖7
You和合作者[13]報道了一起以萘酚和炔烴為底物,Rh催化的經由C(sp2)-H鍵活化的不對稱脫芳構化反應(圖8)。研究發現,手性Rh催化系統可催化萘酚化合物C(sp2)-H活化,與炔烴環合生成螺環分子結構,產物ee值最高可達94%。該反應應用前景廣,可用于構建含有手性螺環結構化合物。

圖8
2016年,Nishimura等人[14]報道了一起以Ir/BINAP為催化體系,以內酰胺化合物和炔烴化合物為底物,經過 C(sp2)-H 鍵活化和不對稱[3+2]成環反應生成螺氨基茚化合物的反應(圖9-a)。該反應產物的對映選擇性可通過添加酸進行改變,在復雜生物活性化合物的合成方面具有很高的應用價值。同年,該課題組[15]以乙烯醚類化合物為烷基化試劑,開發了一種Ir催化的通過C(sp2)-H鍵活化的N-甲磺酰基苯甲酰胺的不對稱烷基化反應(圖9-b)。該反應底物適應性和支鏈選擇性好,產物收率高,ee值最高可達96%。2017年,該課題組[16]通過調節過渡金屬-手性配體催化體系,又成功地將該芳基底物拓展至2-芳基唑類化合物(圖 9-c)。

圖9
2016 年,Lautens等人[17]利用一鍋法,以 α-重氮酰胺化合物為底物,在雙過渡金屬(Pd和Ru)催化下,經過不對稱烯丙基化烷基化反應,成功合成了具有季碳手性中心的3-烯丙基-3-芳基氧化吲哚化合物 (圖10)。該反應產率最高可達99%,ee值最高可達85%。經研究發現,配體的選擇對該連續化反應十分重要。

圖10
2 總結與展望
綜上所述,利用過渡金屬催化 C(sp2)-H鍵活化反應構建手性碳原子中心已經得到了研究人員的廣泛關注,目前已經有大量關于此類反應的文獻報道。因此,如何合適的過渡金屬-手性配體催化體系,實現立體選擇性的碳氫鍵活化,構建手性碳原子中心,正得到越來越深入的研究。該類反應在不對稱合成反應中,也會得到更加廣泛的應用。
但是,雖然如此,過渡金屬催化 C(sp2)-H 鍵活化反應構建手性碳原子中心的反應仍具有其不足之處。具體表現在:(1)反應底物適應性不強,特定催化體系只能應用于特定反應之中;(2)反應規模小,很少有反應能達到克級,能夠應用于工業生產的反應微乎其微;(3)有些反應的反應機理并未得到清楚闡述,這對該類反應的進一步研究和應用有很大的不便之處。
因此,在該領域的探索和研究過程中,我們不能滿足于目前該類反應在實驗室里所取得的飛速進展,更要不斷深入研究,積極驗證,嘗試和構建該類反應的新思路和新技巧,為該類反應從學術界走向工業界提供切實可靠的理論基礎和生產方案。
參考文獻:
[1] (a)Uenishi J,Beau J M,Armstrong R W,et al.Dramatic rate enhancement of suzuki diene synthesis:Its application to palytoxin synthesis[J].J.Am.Chem.Soc., 1987, 109(15): 4756-4758. (b) Lee M M, Gao Z, Peterson B R.Synthesis of a fluorescent analogue of paclitaxel that selectively bindsmicrotubulesand sensitively detects efflux by P-glycoprotein[J].Angew.Chem.Int.Ed., 2017,56(24): 6927-6931.
[2] (a)Clavier H,Boulanger L,Audic N,et al.Design and synthesis of imidazolinium salts derived from (L)-valine.Investigation oftheir potentialin chiralmolecular recognition[J].Chem.Commun., 2004, (10): 1224-1225.(b) Ma D W, Yao J C.Synthesis of chiral N-aryl-αamino acids by Pd-Cu catalyzed couplings of chiral αamino acids with aryl halides[J].Tetrahedron Asymmetry,1996, 7(11): 3075-3078.
[3] (a)Ye B H,Cramer N.Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C-H functionalization[J].Science, 2012.338(6106): 504-506. (b) Wencel-Delord J, Colobert F.Asymmetric C (sp2)-H activation[J].Chem.Eur.J., 2013, 19(42):14010-14017.
[4] Song G Y,Wylie W N O,Hou Z M.Enantioselective CH bond addition of pyridines to alkenes catalyzed by chiral half-sandwich rare-earth complexes[J].J.Am.Chem.Soc., 2014, 136(35): 12209-12212.
[5] Chen D F,Zhao F,Hu Y,et al.C-H Functionalization/asymmetric Michael addition ca scade enabled by relay catalysis:metal carbenoid used for C-C bond formation[J].Angew.Chem.Int.Ed., 2014, 53(40): 10763-10767.
[6]Jia S,Xing D,Zhang D,et al.Catalytic asymmetric functionalization of aromatic C-H bonds by electrophilic trapping of metal-carbene-induced zwitterionic intermediates[J].Angew.Chem., Int.Ed., 2014, 53 (48): 13098-13101.
[7] Xu B,Li M L,Zuo X D,et al.Catalytic asymmetric arylation of α aryl-α-diazoacetates with aniline derivatives[J].J.Am.Chem.Soc., 2015, 137(27): 8700-8703.
[8] Xiao K J, Lin D W,Miura M, et al.Palladium (II)-catalyzed enantioselective C(sp3)-H activation using a chiral hydroxamic acid ligand[J].J.Am.Chem.Soc., 2014, 136(22), 8138-8142.
[9]Shibata T,Ryu N,Takano H.Very important publication:iridium-catalyzed intramolecular enantioselective C-H alkylation at the C-2 position of N-alkenylindoles[J].Adv.Synth.Catal., 2015, 357(6): 1131-1135.
[10]Lee P S,Yoshikai N.Cobalt-catalyzed enantioselective directed C-H alkylation of indole with styrenes[J].Org.Lett., 2015, 17(1): 22-25.
[11]Shirai T,Yamamoto Y.Cationic iridium/S-Me-BIPAM-catalyzed direct asymmetric intermolecular hydroarylation of bicycloalkenes[J].Angew.Chem.Int.Ed., 2015, 54(34): 9894-9897.
[12] (a) Aufdenblatten R, Diezi S, Togni A.Iridium(I)-catalyzed asymmetric intermolecular hydroarylation of norbornene with benzamide[J].Monatsheftefür Chemie., 2000,131:1345-1350. (b)Dorta R,Togni A.Addition of the ortho-C-H bonds of phenol across an olefin catalysed by a chiral iridium(I) diphosphine complex[J].Chem.Commun., 2003, (6): 760-761.
[13]Zheng J,Wang S B,Zheng C,et al.Asymmetric dearomatization of naphthols via a Rh-catalyzed C(sp2)-H functionalization/annulation reaction[J].J.Am.Chem.Soc.,2015, 137(15): 4880-4883.
[14]Nagamoto M,Yamauchi D,Nishimura T.Iridium-catalyzed asymmetric [3+2]annulation of aromatic ketimines with alkynes via C-H activation:unexpected inversion of the enantioselectivity induced by protic acids[J].Chem.Commun., 2016,52(34): 5876-5879.
[15]Hatano M,Ebe Y,Nishimura T,et al.Asymmetric alkylation of N sulfonylbenzamides with vinyl ethers via C-H bond activation catalyzed by hydroxoiridium/chiral diene complexes[J].J.Am.Chem.Soc., 2016, 138(12): 4010-4013.
[16]Yamauchi D,Nishimura T,Yorimitsu H.Asymmetric hydroarylation of vinyl ethers catalyzed by a hydroxoiridium complex:azoles as effective directing groups[J].Chem.Commun., 2017, 53(18): 2760-2763.
[17]Yamamoto K,Qureshi Z,Tsoung J,et al.Combining Rucatalyzed C-H functionalization with Pd-catalyzed asymmetric allylic alkylation:synthesis of 3 allyl-3-aryl oxindole derivatives from aryl α diazoamides[J].Org.Lett.,2016, 18(19): 4954-4957.
