餐飲食品中罌粟殼的快速測定方法
2018-04-19 03:30:10丁建蘭黃恩堂
現代食品 2018年24期
◎ 黃 寧,丁建蘭,黃恩堂,楊 希
(鷹潭市綜合檢驗檢測中心,江西 鷹潭 335000)
近年來,人們越來越關注食品中非法添加藥物的問題。黨的十九大報告明確指出“實施食品安全戰略,讓人民吃得放心”。中央已經把食品安全提高到戰略高度,但在食品中非法添加罌粟殼來吸引顧客的行為,仍屢禁不止。長期食用含罌粟殼的食品會對人的神經系統、消化系統、內分泌系統造成危害。為了維護廣大人民群眾的身體健康,要嚴厲打擊非法添加現象,建立快速、準確的餐飲食品中罌粟殼的測定方法具有重要意義。
餐飲食品中罌粟殼的檢測方法于2018 年之前尚無國家標準,2018 年4 月市場監管總局發布的《食品中罌粟堿、嗎啡、可待因、那可丁和蒂巴因的測定液相色譜-串聯質譜法》補充了檢驗方法,由于設備資源有限,基層推廣時無法直接照搬使用,給我國餐飲食品的監督管理工作帶來了一定的技術難題,為進一步保障人民的飲食安全,維護人民身體健康,研究餐飲食品中罌粟殼的檢測方法勢在必行。筆者參考了大量文獻[1-67],在相關研究的基礎上,建立了一種經濟、快速、準確且靈敏度高的餐飲食品中罌粟殼的檢測方法。
1 儀器與材料
1.1 儀器
液相色譜/質譜聯用儀(三重串聯四級桿質譜)LC/MS QQQ(配四元泵、柱溫箱、自動進樣器、電噴霧離子源ESI,美國Agilent Technologies 1290 Inf inity Ⅱ/6460 Triple Quad LC/MS);電子天平(賽多利斯科學儀器上海有限公司MSA225P-CE)。
1.2 材料
鹽酸、乙醇、正丁醇、三氯甲烷、濃氨、碘化鉍鉀試液、碘化汞鉀試液、甲醛硫酸試液、鉬硫酸試液、稀鐵氰化鉀試液、氫氧化鈉、乙酸乙酯、甲酸、丙酮、氨水、苯、甲苯及亞硝酸鈉乙醇試液為分析純;甲醇、乙腈為色譜純,水為超純水。過濾用0.22 μm 濾膜。其他材料及其來源見表1 和表2。

表1 罌粟殼藥材來源表
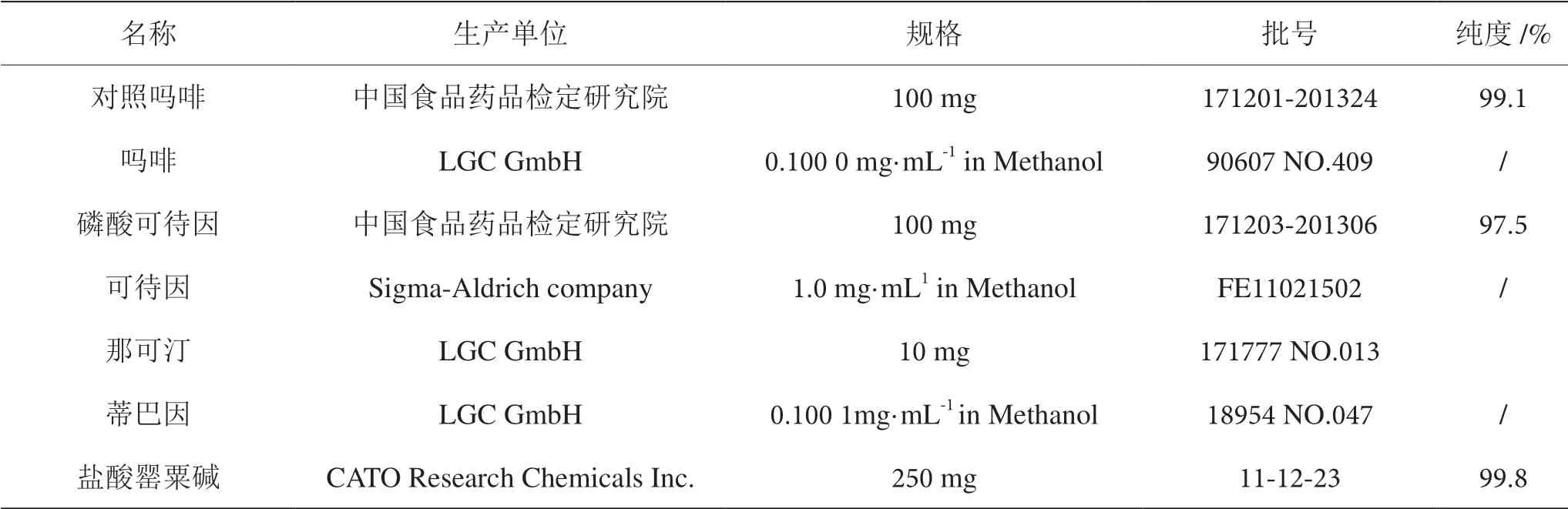
表2 標準品來源表
待測樣品為市場上的牛肉粉面、烤肉串、醬鹵雞腿、 火鍋底料、自制醬料及麻辣燙6 類食品共25 批樣品。
2 試驗條件的選擇
2.1 理化反應試劑的選擇
根據生物堿特性,分別用碘化鉍鉀試液、碘化汞鉀試液、甲醛硫酸試液、鉬硫酸試液和稀鐵氰化鉀試液與樣品反應,結果顯示(圖1)加碘化鉍鉀試液(管1、3)、碘化汞鉀試液(管2、4)反應效果好,陽性對照與陰性對照結果區別很明顯。

圖1 化學鑒別反應結果圖
2.2 薄層色譜條件的選擇
2.2.1 薄層板的選擇
分別用硅膠G 薄層板、硅膠GF254薄層板、2%氫氧化鈉溶液制備的硅膠G薄層板試驗。
2.2.2 展開劑的選擇
分別用乙酸乙酯-甲酸-氨水(13 ∶2 ∶1)、三氯甲烷-丙酮-氨水(16 ∶4 ∶0.3)、三氯甲烷-甲醇-氨水(13 ∶2 ∶0.3)、丙酮-苯-乙醇-氨水(20 ∶20 ∶5 ∶2)、乙酸乙酯-甲醇-氨水(85 ∶5 ∶2)以及甲苯-丙酮-乙醇-濃氨試液(20 ∶20 ∶3 ∶1)作為展開劑進行試驗。
2.2.3 顯色劑的選擇
分別用碘化鉍鉀試液、亞硝酸鈉乙醇試液、改良碘化鉍鉀試液噴在展開后的薄層板上。
2.2.4 檢視條件的選擇
分別在日光下、紫外光燈254 nm 下、紫外光燈365 nm 下檢視。
2.2.5 結果
檢視結果如圖2 ~4 所示。

圖2 噴顯色劑碘化鉍鉀 試液后日光下檢視圖

圖3 紫外光燈365 nm 下 檢視圖

圖4 噴顯色劑亞硝酸鈉乙醇試液后日光下檢視圖
如圖3 所示,薄層板用硅膠G 板較好,選擇“丙酮-苯-乙醇-氨水(20 ∶20 ∶5 ∶2)”作為展開劑,5 種生物堿分離效果好,生物堿對照品點1 ~5 分離較好。顯色劑選擇碘化鉍鉀試液較理想,目標物質斑點顯色清晰,呈橙紅色,薄層板背景呈淡黃色,顏色均勻。如圖3 中點5 所示,噴顯色劑亞硝酸鈉乙醇試液后,目標物質的斑點明顯變成鮮艷的橘紅色,斑點顏色明顯,但界限不清晰,而且薄層板背景由深紅棕色慢慢轉白,顏色不均勻。
檢視在日光下較好,如圖2 所示。在紫外光365 nm下檢視很不理想,目標物質沒有熒光斑點,檢視不出來,如圖4 中點1 ~5 所示;樣品中的其他雜質熒光斑點清晰,如圖4 中點7 所示。
2.3 流動相的選擇
試驗嘗試多種流動相:甲醇-0.02 mol·L-1乙酸銨溶液、乙腈-0.02 mol·L-1磷酸二氫鉀+0.02 mol·L-1磷酸氫二鈉氫溶液、乙腈-0.01mol·L-1乙酸銨溶液(pH3.0、3.5、3.8、4.0、4.3),試驗表明乙腈-0.01 mol·L-1乙酸銨溶液(pH4.3)梯度洗脫分離效果最好,如圖5 所示。
2.4 柱溫的選擇
分別在30、35、40 ℃柱溫下進行試驗,結果在柱溫40 ℃時峰分離度和峰型最好,如圖5 所示。
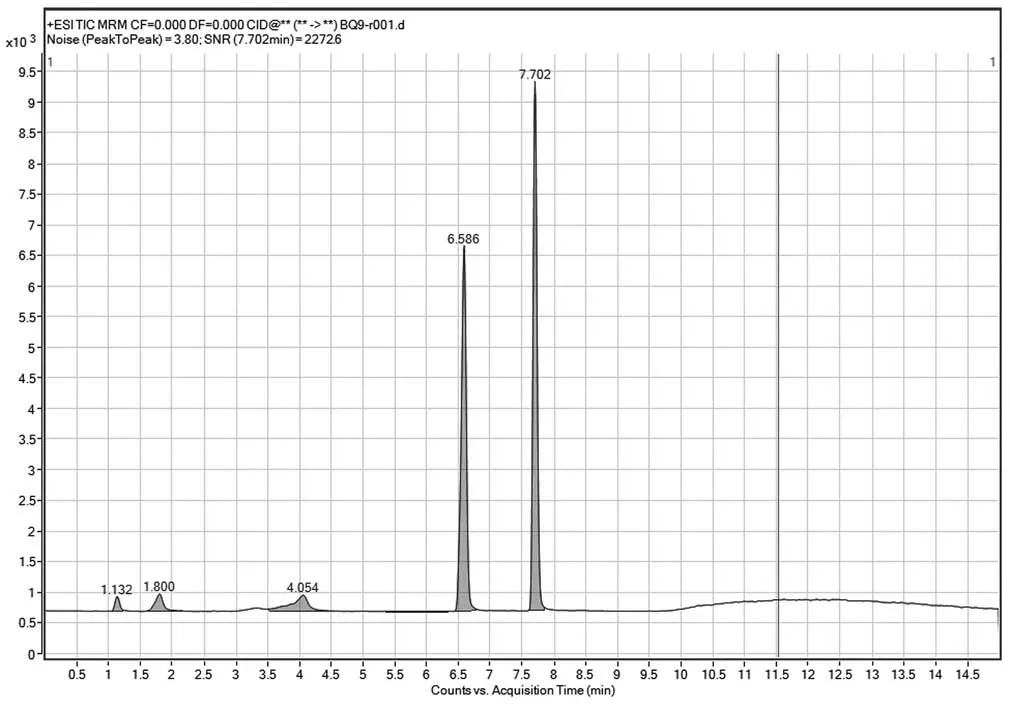
圖5 40 ℃柱溫時峰分離度圖
3 方法與結果
3.1 理化反應法
3.1.1 方法
取樣品5 g,加5%鹽酸乙醇溶液20 mL,超聲 30 min,趁熱濾過,濾液蒸至近干,殘渣中加5%鹽酸溶液5 mL 使之溶解,分置二支試管中,一管中加碘化鉍鉀試液,生成橙紅色沉淀;另一管中加碘化汞鉀試液,生成灰白色沉淀。
3.1.2 結果判斷
供試品應不呈正反應,若兩管均呈正反應,則結果疑似陽性,待液-質聯用法確證。
3.2 薄層色譜法
3.2.1 實驗方法
(1)對照品溶液的制備。分別取嗎啡、那可汀、可待因、蒂巴因及罌粟堿對照品,用甲醇分別稀釋至0.2、0.2、0.4、0.8 mg·mL-1與0.4 mg·mL-1的溶液,即得單個對照品溶液。將5 種對照品溶于同一容量瓶中,得混合對照品溶液。
(2)樣品溶液的制備。取樣品10 g,加甲醇30 mL 加熱回流30 min,趁熱濾過,濾液蒸至近干,殘渣加甲醇1 mL 溶解。
(3)薄層板。硅膠G 板。
(4) 展 開 劑。 丙 酮∶苯∶乙 醇∶氨 水(20 ∶20 ∶5 ∶2)。
(5)檢視。噴碘化鉍鉀顯色劑,標準品與陽性樣品應出現明顯的橙紅色斑點。
3.2.2 結果判斷
供試品色譜中,在與對照品色譜相應的位置上,不得顯相同顏色的斑點。若顯相同顏色的斑點,則為疑似陽性,待液-質聯用法確證。
3.3 液-質聯用法
3.3.1 標準曲線的制備
分別精密稱取嗎啡、那可汀、可待因、蒂巴因及罌粟堿對照品,嗎啡用甲醇稀釋成系列濃度40、50、100、200、300、400 ng·mL-1及500 ng·mL-1,可待因用甲醇稀釋成系列濃度40、50、100、200、300、400、500、1 000 ng·mL-1及2 500 ng·mL-1,罌粟堿、蒂巴因和那可汀用甲醇稀釋成系列濃度2、4、8、10、20、40、60、80 ng·mL-1及100 ng·mL-1,分別精密吸取標準溶液各5μL,注入液-質聯用儀分析后得到不同濃度的峰面積,以峰面積為縱坐標,罌粟殼生物堿濃度 (ng·mL-1)為橫坐標,分別進行線性回歸分析。
3.3.2 樣品溶液的制備
稱取樣品5 g(精確至0.01 g)于50 mL 聚四氟乙烯具塞離心管中,加水5 mL,振搖使之分散均勻(有些不易搖散的樣品,必要時可加水10 mL),加入乙腈15 mL,渦旋振蕩1 min,加入6 g 無水硫酸鎂和1.5 g 無水乙酸鈉的混合粉末,迅速振搖。渦旋振蕩 1 min,以4 000 r·min-1離心5 min,精密量取上清液 10 mL,用經活化的C18小柱凈化,用10 mL 甲醇洗脫,收集洗脫液,氮吹至干,用甲醇定容至2 mL,經 0.22 μm 濾膜過濾,待測。
3.3.3 色譜條件
以十八烷基硅烷鍵合硅膠為色譜柱填充劑(規格為2.1 mm×100 mm,5 μm),柱溫40 ℃,流速 0.2 mL·min-1,檢測波長:250 nm,掃描波長范圍 200 ~400 nm,進樣量5 μL,以乙腈為流動相A,0.01 mol·L-1乙酸銨溶液(pH4.3)為流動相B,按表3進行梯度洗脫。
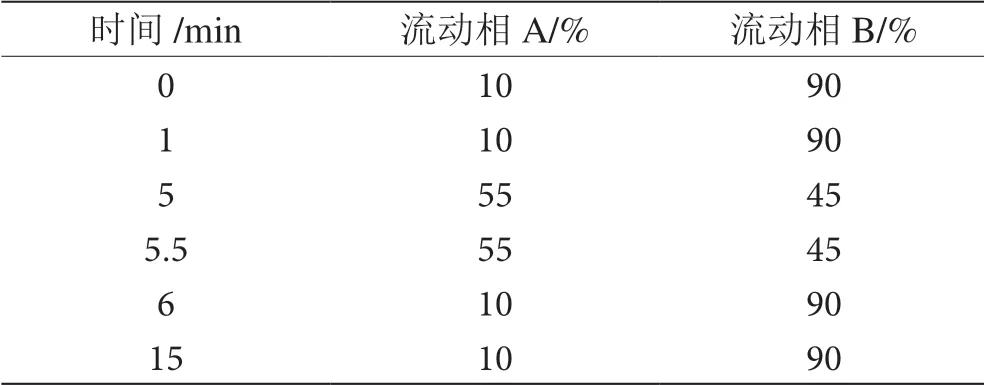
表3 洗脫條件表
3.3.4 質譜條件ESI 離子源干燥氣溫度300 ℃,干燥氣流速11 L·min-1,毛細管電壓4 000 V,正掃描模式,MRM 多反應監測,待測化合物監測母離子、子離子、碎裂電壓及碰撞能等質譜參數見表4。
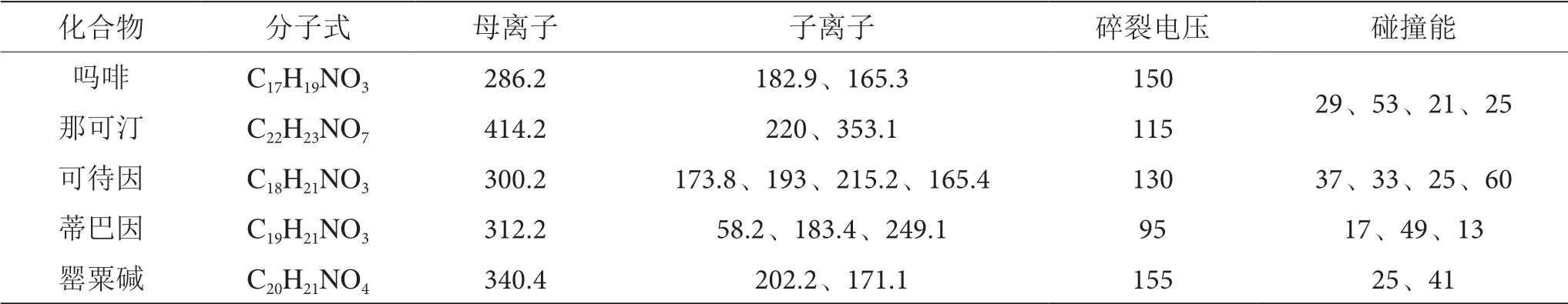
表4 待測化合物的質譜參數表
3.3.5 測定法
分別精密吸取供試品溶液和標準曲線溶液各 5 μL,注入液-質聯用儀,記錄色譜圖。
3.3.6 結果判斷
供試品色譜中,應不得出現與標準品色譜保留時間一致的色譜峰。若出現保留時間相同的色譜峰,則相應的一級質譜及二級質譜均不得與對照品一致。若色譜圖、一級質譜及二級質譜均與對照品一致,結果判定為陽性。陽性結果按式(1)定量計算。
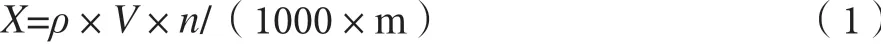
式(1)中:X—試樣中待測組分的含量,單位為mg·kg-1。
ρ—從標準曲線得到的試樣中待測組分的含量,單位為ng·mL-1。
V—試樣的體積,單位為mL。
m—試樣的質量,單位為g。
n—樣品稀釋倍數。
計算結果保留三位有效數字,在重復性條件下,獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%。
3.3.7 線性關系考察
按2.2.3 做標準曲線,進行線性回歸,嗎啡、可待因、罌粟堿、蒂巴因、那可汀的線性方程及相關系數如表5 所示。

表5 嗎啡、可待因、罌粟堿、蒂巴因、那可汀的回歸方程表
3.3.8 定量限
將已知低濃度的罌粟殼生物堿對照品溶液,按3.3.3 至3.3.6 所述方法測定,以信噪比(S/N)為10 ∶1 時的濃度為定量限(LOQ),嗎啡、可待因、罌粟堿、蒂巴因及那可汀的定量限分別為2.4×10-2、2.4×10-2、6×10-4、6×10-4mg·kg-1及6×10-4mg·kg-1。
3.3.9 精密度
將混合標準品連續進樣6 次,記錄其峰面積,精密度用峰面積變化的相對標準偏差(RSD)表示,嗎啡、可待因、罌粟堿、蒂巴因和那可汀的峰面積的RSD 值分別為1.32%、4.63%、1.11%、0.56%及1.77%,RSD均小于5%,表明精密度良好。
3.3.10 穩定性試驗
在室溫條件下,將同一份樣品溶液每隔一段時間(0、2、4、8 h 及12 h)測定一次,計算嗎啡、可待因、罌粟堿、蒂巴因且那可汀峰面積變化的相對標準偏差(RSD),分別為3.38%、1.38%、4.44%、4.99%及3.60%。說明樣品在12 h 內穩定性好。
3.3.11 回收率試驗
分別精密稱取相同質量的樣品10 份,一份不加標準品,作為測定本底值的樣品。另外9 份分別加入高、中、低3 種濃度的標準品制備供試液,每種濃度重復3 次,計算其樣品加標回收率,嗎啡的平均加標回收率是78.28%~103.62%,可待因的平均加標回收率是80.86%~108.80%,罌粟堿的平均加標回收率是84.21%~101.61%,蒂巴因的平均加標回收率是74.63%~96.40%,那可汀的平均加標回收率是 76.26%~91.08%。表明本法準確度良好。
4 應用
用本法對市場上的牛肉粉面、烤肉串、醬鹵雞腿、火鍋底料、自制醬料及麻辣燙6 類食品共25 批樣品進行測定,結果均未檢出嗎啡、那可汀、可待因、蒂巴因及罌粟堿。方法檢測準確率達100%,經理化反應法、薄層色譜法及液-質聯用法檢測,確實未檢出罌粟殼生物堿。
5 討論
目前大多研究只采用液相色譜-質譜聯用儀測定罌粟殼中的生物堿,但液相色譜-質譜聯用儀價格昂貴,維護費用高,考慮到基層監管部門儀器設備的有限性和監測方法的可行性,筆者建立的方法經濟快捷,定量準確穩定,易于推廣,為保障人民飲食安全,為加強餐飲食品非法添加的監管提供技術支撐。
