阿替美唑不同種屬體外代謝產物鑒定比較及代謝通路研究
2018-05-13 08:30:26楊春苗仇曉燕張天宏張文鵬蘇瑞斌莊笑梅
中國藥理學與毒理學雜志 2018年12期
李 正,高 尤,楊春苗,仇曉燕,張天宏,張文鵬,蘇瑞斌,莊笑梅
(1.軍事科學院軍事醫學研究院毒物藥物研究所,抗毒藥物與毒理學國家重點實驗室,北京100850;2.海軍青島特勤療養中心,山東青島266071)
阿替美唑(atipamezole)是具有咪唑結構的α2-腎上腺素受體拮抗劑[1],能迅速扭轉由α2-腎上腺素受體激動劑誘導的鎮靜和麻醉作用,臨床上常被用來拮抗α2-腎上腺素受體激動劑(尤其是右美托咪定)的麻醉作用,同時還能有效地緩解美托咪定-咪達唑侖-氯胺酮聯合麻醉引起的不良反應[2]。近年來研究發現,阿替美唑還對疼痛具有調節作用,可阻斷去甲腎上腺素能神經通路疼痛傳導途徑,導致疼痛相關反應的增加,但不影響持續性傷害性疼痛的刺激。肌內注射阿替美唑(0.01~0.03 mg·kg-1)可增強雄性猴的性行為,增加射精量及性交次數[3]。基于以上作用,阿替美唑的開發與臨床應用前景相當廣闊,但目前尚未見有關阿替美唑藥物代謝特征研究的報道。
在早期藥物-藥物相互作用研究中,肝微粒體與重組人源細胞色素P450(cytochrome P450,CYP)同工酶體外孵育體系是研究藥物代謝特征的重要體外模型,可用于鑒定代謝產物、研究代謝酶表型和代謝通路等[4]。本研究應用不同種屬肝微粒體以及重組人源CYP同工酶體外溫孵模型獲得體外代謝產物;體外樣品采用Thermo Q-Exactive型超高效液相色譜串聯四極桿-靜電場軌道阱高分辨質譜系統(ultra performance liquid chromatography coupled with linear ion trap-Orbitrap mass spectrometry,UPLC/LTQ-Orbitrap-MS檢測分析[5],通過比對精確相對分子質量和二級碎片離子信息鑒定阿替美唑在不同種屬肝微粒體以及重組人源CYP同工酶中的代謝產物,揭示其代謝轉化的種屬差異以及代謝通路,旨在為新藥的進一步開發和可能的藥物相互作用研究提供科學數據。
1 材料與方法
1.1 藥品和試劑
阿替美唑,白色粉末,批號:S160801,純度99.39%,軍事科學院軍事醫學研究院毒物藥物研究所合成;鹽酸普萘洛爾,白色粉末,批號:318-98-9,純度99.8%,購于(中國)北京百靈威科技有限公司。甲醇和乙腈(色譜純),購于美國Fisher Scientific公司;甲酸購于賽默飛世爾科技(中國)有限公司;還原型輔酶Ⅱ(NADPH),批號:8516010103,購于美國Roche公司。
混合人肝微粒體(150人份,批號:38292)、大鼠肝微粒體(批號:7024004)和小鼠肝微粒體(蛋白濃度20 g·L-1,批號:7352001)購于美國BD Gentest公司。比格犬肝微粒體(蛋白濃度20 g·L-1,批號:1410114)購于美國Sekisui Xenotech公司;猴肝微粒體(蛋白濃度20 g·L-1,批號:NMZC)購于美國IVT公司。
重組人源CYP同工酶rCYP1A2+OR+b5(批號:3107807),rCYP2A6+OR+b5(批號:7116001),rCYP2B6+OR+b5(批號:626605),rCYP2C8+OR+b5(批號:8190001),rCYP2C9+OR+b5(批號:6257001),rCYP2C19+OR+b5(批號:6173002),rCYP2D6+OR+b5(批號:7114001),rCYP3A4+OR+b5(批號:5105002),rCYP2J2+OR+b5(批號:7311001),rCYP2E1+OR+b5(批號:6341001)和rCYP4F2+OR+b5(批號:8050001),購于美國BD Gentest公司(1000 nmol·L-1)。
1.2 主要儀器和設備
FRESCO21型離心機(美國Thermo Fisher公司);XW-80A型漩渦混合器(上海青浦滬西儀器廠);BS110S型電子分析天平(北京賽多利斯天平有限公司);Thermo Scientific Q Exactive質譜聯用儀和Mass Frontier7.0軟件分析系統(美國Thermo Fisher科技公司);UltiMate 3000型超高效液相色譜儀(美國戴安公司)。
1.3 肝微粒體與重組人源CYP同工酶體外溫孵實驗
不同種屬肝微粒體與重組人源CYP同工酶孵育實驗分為3組(n=3),依次為實驗組、陽性對照組和陰性對照組。實驗組為含阿替美唑的孵育體系;陽性對照組為含陽性探針底物(非那西丁、安非他酮、阿莫地奎、雙氯芬酸、S-美芬妥英、右美沙芬和咪達唑侖)的孵育體系,以確定反應體系的活性;陰性對照組的孵育體系中不含NADPH,以確定阿替美唑在反應體系中的穩定性。
1.3.1 肝微粒體孵育實驗
不同種屬(大鼠、比格犬和人)肝微粒體溶液中加入阿替美唑,置37℃水浴預孵育5 min后,加入NADPH啟動反應,孵育體系中各成分終濃度為:阿替美唑50 μmol·L-1,肝微粒體蛋白1 g·L-1,NADPH 1 mmol·L-1,總孵育體積為800 μL。分別于加入NADPH啟動反應后0和60 min加入2倍體積冰冷的含普萘洛爾(內標)30 mg·L-1的乙腈溶液終止反應。將終止反應的孵育樣品渦旋振蕩2 min后,4℃下13 800×g離心10 min,將上清液全部取至1.5 mL離心管,用氮氣吹干后用體積200 μL的流動相復溶,即為待檢樣品。
1.3.2 重組人源CYP同工酶孵育實驗
各重組人源同工酶(rCYP1A2,rCYP2B6,rCYP2C8,rCYP2C9,rCYP2C19,rCYP2D6,rCYP3A4和rCYP2A6)加入阿替美唑,置37℃水浴預孵育5 min后,加入NADPH啟動反應,反應體系中各成分終濃度為:阿替美唑50 μmol·L-1,重組酶濃度20 nmol·L-1,NADPH 1mmol·L-1,總體積800 μL。終止反應及制備待檢樣品的條件與步驟同1.3.1。
1.4 UPLC/LTQ-Orbitrap-MS分析條件
色譜條件:色譜柱為Phenomenex C1(8100 mm×2.1 mm,5 μm)柱;A相:0.1%甲酸水,B相0.1%甲酸乙腈;采用梯度洗脫(0~1 min,2%B;1~15 min,2%~95%B;15~17 min,95%B;17~20 min,2%B);流速0.5 mL·min-1;進樣量5 μL。
質譜條件:質譜采用電噴霧離子化源(electrospray ionization,ESI)正離子電離模式;霧化溫度300℃;離子傳輸管溫度325℃;鞘氣流速40 arb;輔助氣流速10 arb;噴霧電壓3.5 kV;碰撞電壓20,40和60 eV;質荷比(mass-to-charge ratio,m/z)檢測范圍50~750;正離子檢測模式;采用高分辨全離子掃描模式;一級全離子掃描(分辨率R=70 000,掃描質量范圍m/z 50~750),二級全離子掃描(分辨率R=17500)。
1.5 數據分析處理
高分辨全離子掃描模式下的總離子流色譜(total ion chromatogram,TIC)圖,運用MetWorksTM1.3 SP4軟件進行多重質量虧損處理(multiple mass defect filters),發現新生成的代謝產物。利用Xcaliber4.0工作站,根據精確分子質量數據推測可能的元素組成,并結合質譜裂解碎片信息進行數據處理,采用分子式預測模塊預測所有母離子和碎片離子的分子式,鑒定阿替美唑的體外代謝產物結構。
2 結果
2.1 各種屬肝微粒體阿替美唑代謝產物譜比較
通過對比各種屬肝微粒孵育實驗中0和60 min的TIC圖發現,阿替美唑的保留時間為6.5 min,經肝微粒體代謝酶體外代謝后出現3個產物峰(圖1),其色譜行為一致,保留時間分別為4.9,5.4和8.0 min,依次命名為M1,M2和M3,其中M1和M2保留時間早于原型藥物,M3的保留時間落后于原型藥物。各種屬肝微粒體中3個產物的響應強度不同,提示阿替美唑的代謝產物譜無種屬差異,但產物生成比例有一定種屬差異。
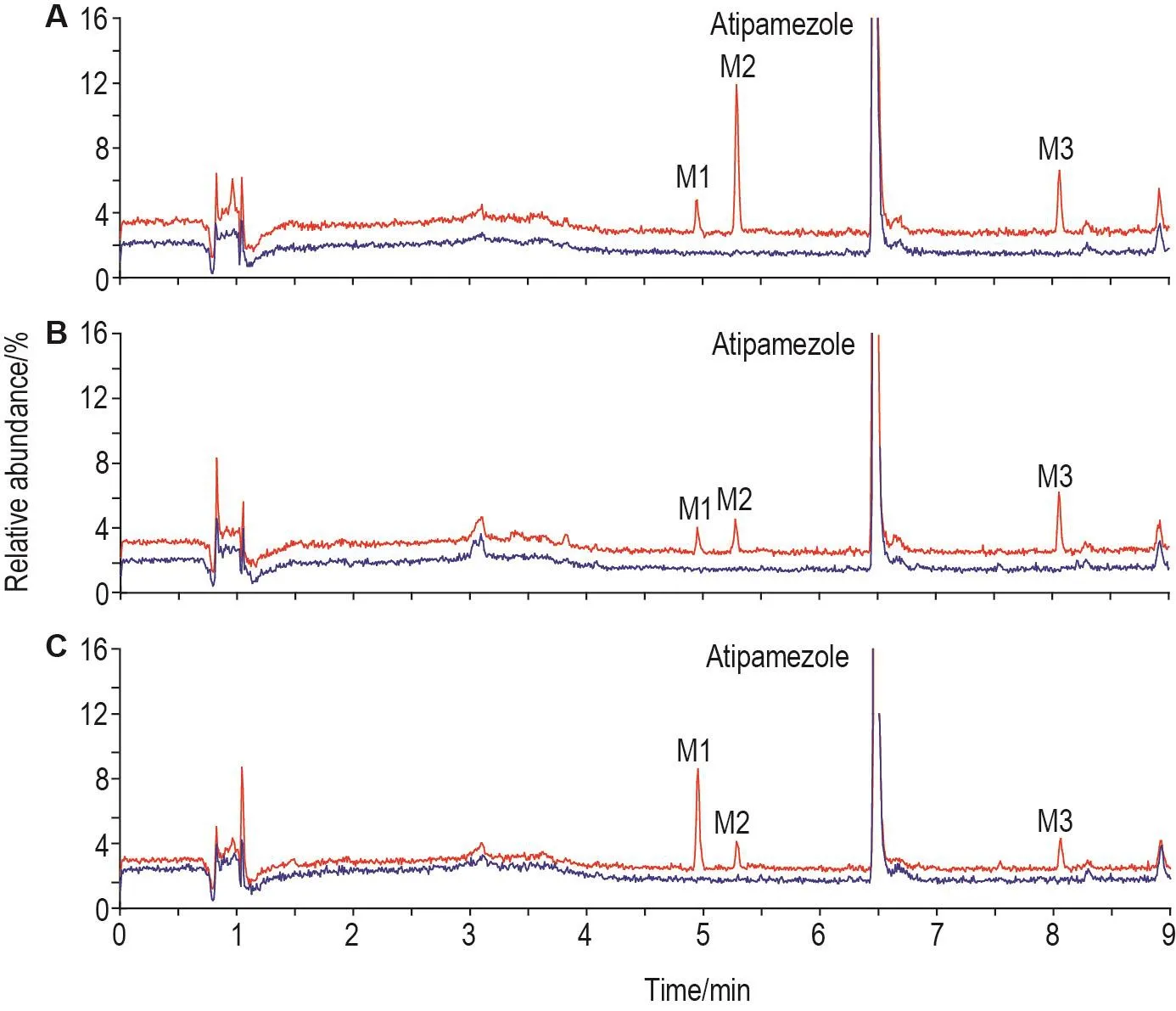
Fig.1 Comparative total ion chromatograms(TlCs)of atipamezole and its metabolites obtained from human(A),dog(B)and rat(C)liver microsomes before and after incubation.Blue curves represent incubation information obtained at 0 min,red curves represent incubation information obtained at 60 min.M1 is the metabolite of terminal ethyl hydroxylation,M2 is the metabolite of benzene epoxidation and M3 is the metabolite of imidazole epoxidation.
2.2 阿替美唑肝微粒體體外代謝產物鑒定
阿替美唑及其3個體外代謝產物的精確分子質量與二級質譜圖見圖2。圖2A中包含阿替美唑和3個代謝產物的母離子和子離子的碎片結構解析;圖2B為阿替美唑的質譜裂解綜合圖,數據見表1。由圖1和表1可知,在正離子模式下,阿替美唑準分子離子峰[M+H]+為m/z 213.13786,經誘導碰撞解離(collision-induced dissociation,CID)裂解后,特征二級碎片離子包括m/z 69.04527,117.08976,145.10071和183.09106。其中碎片離子m/z183.09106為脫乙基的基峰,碎片離子m/z 145.10071為脫咪唑基峰,碎片離子m/z 117.08976為同時脫乙基和咪唑環后的苯并環戊基基峰,碎片離子m/z 69.04527為咪唑環基峰。
代謝產物M1的保留時間為4.9 min,準分子離子峰[M+H]+為m/z 229.13286,比阿替美唑[M+H]+多16 u,根據精確相對分子質量,分子式為C14H16ON2,推測可能為阿替美唑氧化產物。M1的二級特征碎片離子包括m/z 81.04514,95.04914,143.08505和211.12239。由特征碎片離子m/z 211.12239推測該碎片可能為M1脫水峰,因此M1的氧化位點很可能位于乙基,結合氧化代謝性質推測乙基末端發生羥基化的可能性較大,特征碎片離子m/z 143.08505為m/z 211.12239脫去咪唑環后末端形成乙烯結構的基峰。由特征碎片離子m/z 95.04914推測,M1為脫水后形成環戊烯結構后環戊烯開環形成咪唑環加乙烯結構。特征碎片離子m/z 81.04514為m/z 95.04914再脫去甲基形成。
代謝產物M2保留時間為5.3 min,準分子離子峰[M+H]+為m/z 229.13290,比阿替美唑[M+H]+峰多16 u,根據精確相對分子質量,分子式為C14H16ON2,推測M2也可能為阿替美唑氧化產物。M2的二級特征碎片離子包括m/z 69.04521,133.06451,161.09663和199.08592。由特征碎片離子m/z 69.04521推斷M2的咪唑環結構不變;而M2的特征碎片離子m/z 133.06451和161.09663均比阿替美唑特征碎片離子m/z 117.08976和145.10071多16 u,推測M2氧化位點很可能位于苯環處;特征碎片離子m/z 199.08592為M2脫乙基形成的基峰。
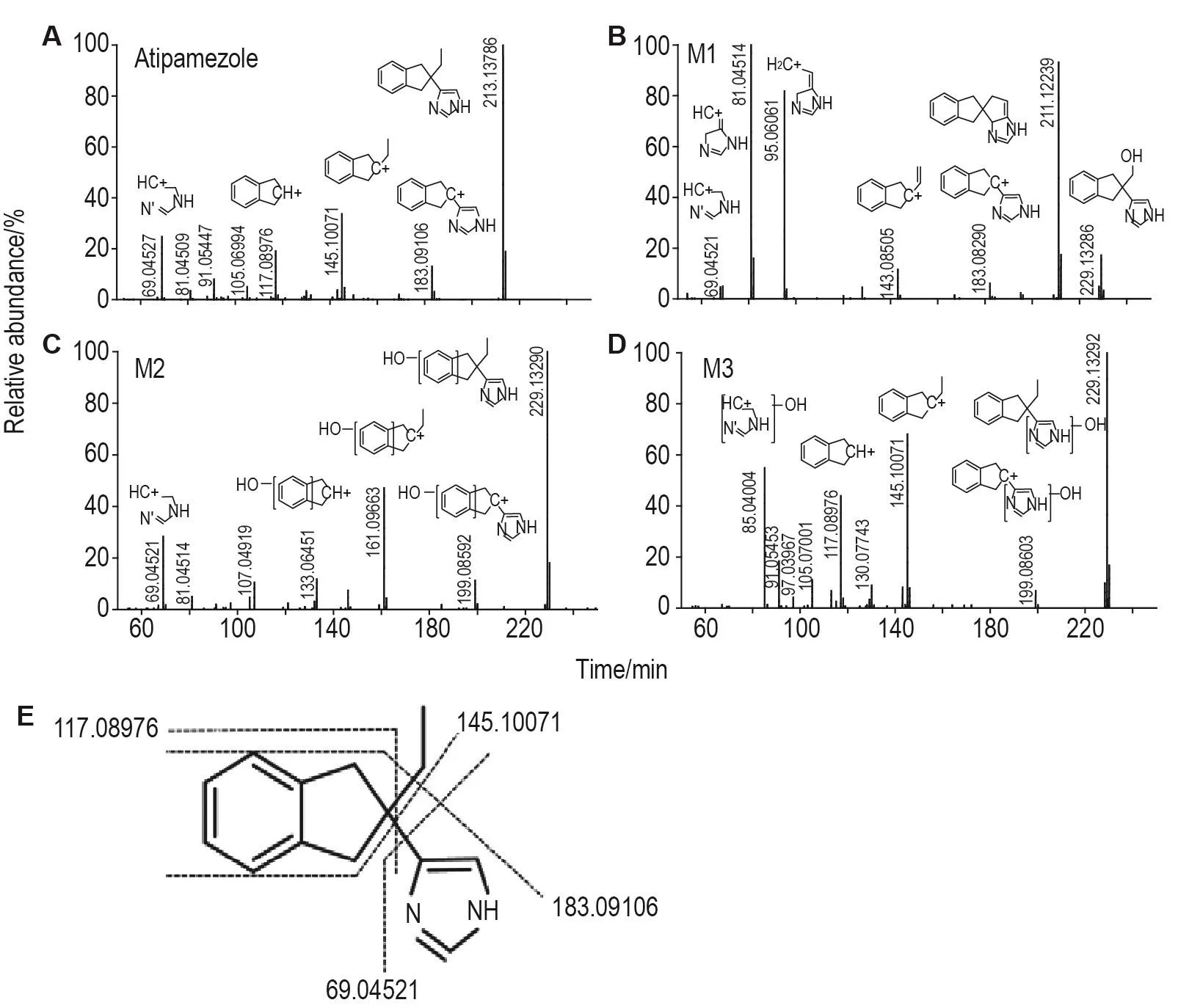
Fig.2 Tandem mass spectrometry(MS/MS)spectra of atipamezole(A),and its metabolites M1(B),M2(C)and M3(D)in human liver microsomes and proposed fragmentation patterns of atipamezole(E).
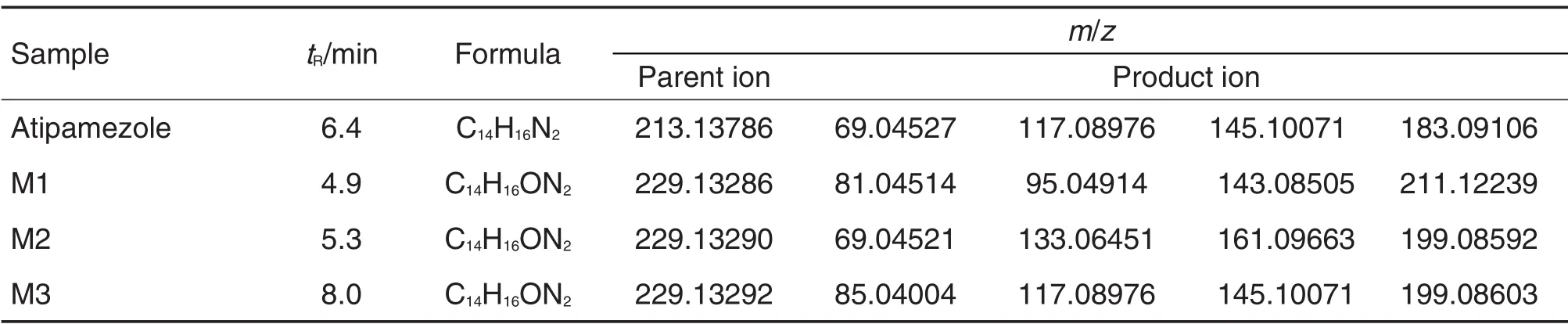
Tab.1 Mass-to-charge ratio(m/z)of atipamezole and its major metabolites after incubation in liver microsomes
代謝產物M3保留時間為8.0 min,準分子離子峰[M+H]+為m/z 229.13292,比阿替美唑[M+H]+峰多16 u,根據精確相對分子質量,分子式為C14H16ON2,推測M3也可能為阿替美唑氧化產物。M3的特征碎片離子包括m/z 85.04004,117.08976,145.10071和199.08603。由特征碎片離子m/z 117.08976,145.10071與阿替美唑的二級特征碎片離子一致推斷M3的苯環結構不變;由特征碎片離子m/z 85.04004比阿替美唑咪唑基峰m/z 69.04527增加16u,推測該碎片可能為咪唑環氧化的基峰。因此,M3氧化位點可能位于咪唑環處,特征碎片離子m/z199.08592為M3脫去乙基形成的基峰。
2.3 阿替美唑在各重組人源CYP同工酶中的代謝轉化
在對比阿替美唑在各重組人源CYP同工酶孵育樣品中0和60 min的TIC圖中選擇提取離子m/z 229.13286得到圖3。結果顯示,除在CYP2A6,2B6,2D6,2C19和3A4外,阿替美唑在其他3個主要同工酶(CYP1A2,2C8和2C9)中不發生代謝轉化,并且不同重組人源CYP同工酶對應不同的代謝產物。M1由CYP2A6,CYP2D6和CYP3A4介導產生,M2由CYP2A6,CYP2D6和CYP2C19介導產生,M3由CYP2A6,CYP2B6和CYP3A4產生。綜上,阿替美唑在肝重組人源CYP同工酶中的代謝通路如圖4。
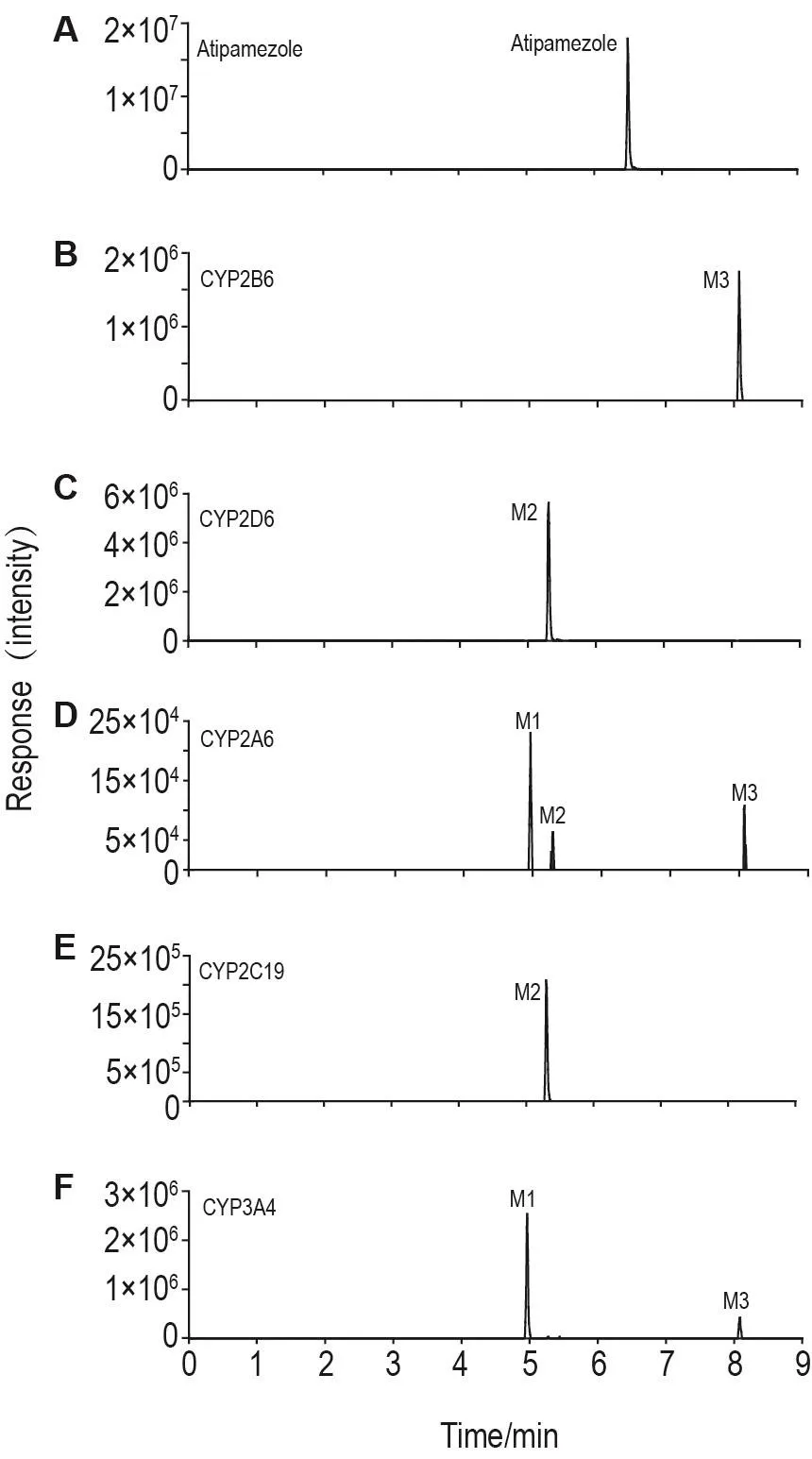
Fig.3 Extracted ion chromatograms of the major metabolites of atipamezole in recombinant human cytochrome P450 enzymes(CYPs)
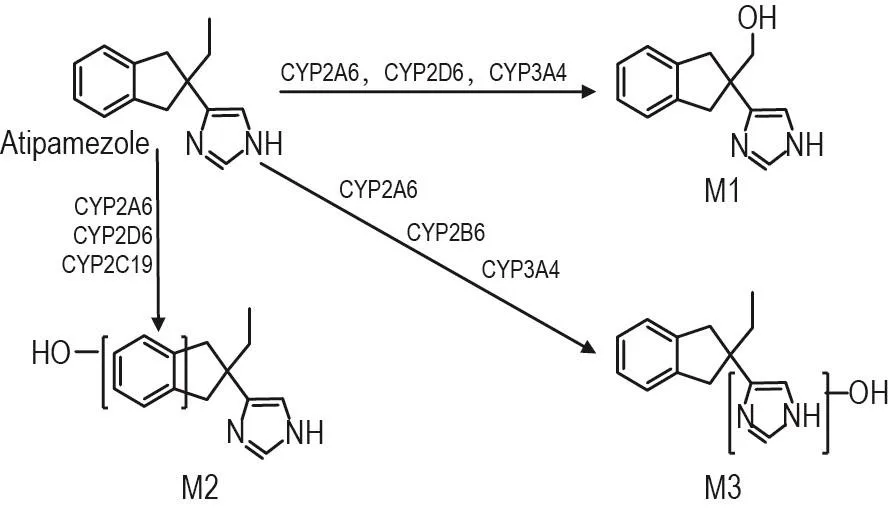
Fig.4 Supposed metabolic pathway of atipamezole in recombinant human CYPs.
3 討論
在新藥非臨床評價中,動物種屬的選擇非常重要。根據《FDA新藥安全性評價指南》要求[6],化合物在非臨床受試動物中的代謝產物譜盡量與人體代謝產物譜一致,或不遺漏人體代謝產物,才能保證非臨床安全性評價結果的可靠性及臨床相關性。因此,新藥的代謝產物鑒定和產物譜的種屬比較是非臨床藥動學及安全性評價的重要研究內容[7]。
3.1 阿替美唑Ⅰ相代謝產物譜的種屬比較
不同種屬體外肝微粒孵育實驗是獲得主要Ⅰ相代謝產物的重要手段,可以快速鑒定代謝產物并比較不同種屬產物譜[8]。本研究基于靈敏的測定肝微粒孵育體系中阿替美唑及其代謝產物的UPLC/LTQ-Orbitrap-MS方法,發現阿替美唑經大鼠、比格犬和人肝微粒體孵育后都產生了3個主要代謝產物,特別是人肝微粒體孵育后產生的代謝產物在大鼠和比格犬中均有產生,提示大鼠和比格犬可作為非臨床藥物安全性評價的動物種屬。雖各種屬中生成3個產物的質譜響應不同,但由于缺乏產物標準品,且質譜檢測對基質的敏感性較高[6],無法準確比較產物生成量。
3.2 阿替美唑Ⅰ相代謝產物初步鑒定
四極桿-靜電場軌道阱高分辨質譜通過測定各離子的振蕩頻率,運用傅里葉變換計算m/z,其分辨率可達10萬。采用精確質量數提取獲得相應化合物的質譜響應,充分保證了方法的高選擇性[9]。本研究采用UPLC/LTQ-Orbitrap-MS技術,基于多重質量虧損方法,在20 min內對鹽酸阿替美唑的3個代謝產物進行了分離和鑒定。阿替美唑3個Ⅰ相代謝產物均為氧化產物,根據母藥的質譜裂解規律,結合精確分子質量數據推測可能的元素組成,逐一解析了產物的氧化代謝位點,包括乙基末端羥基化、苯環羥基化及咪唑環氧化。
3.3 阿替美唑的Ⅰ相代謝通路
應用重組人源同工酶孵育后進一步明確了阿替美唑在各同工酶中的代謝轉歸。阿替美唑在CYP2A6,2B6,2D6,2C19和3A4中轉化成不同的代謝產物(圖4)。Ⅰ相代謝通路研究結果顯示,CYP2A6,2B6,2D6,2C19和3A4均參與了鹽酸阿替美唑的氧化代謝,并且每個代謝產物都是有多個代謝酶亞型介導的。CYP2A6,2D6和2C19雖均有明顯的基因多態性[10-11],但由于阿替美唑的多通路代謝,這種因素的影響可能較小。
綜上,本研究首次鑒定了阿替美唑的3個體外氧化產物,明確了參與Ⅰ相代謝的代謝酶亞型。由于阿替美唑代謝通路廣泛,在臨床與其他藥物合用時可能不易發生作為CYP藥物代謝酶底物介導產生的藥物-藥物相互作用[12-14]。在后期研究中還需要進一步明確阿替美唑對藥物代謝酶的抑制和誘導作用,以評價基于代謝酶調節作用產生的藥物相互作用。
