超高效液相色譜-串聯質譜法測定化妝品中6種生物堿
2018-07-06 01:53:26汪晨霞張瑞瑞尋知慶郭新東黃金鳳
分析測試學報 2018年6期
汪晨霞,張瑞瑞,尋知慶,郭新東,王 強,黃金鳳
(廣州質量監督檢測研究院,廣東 廣州 511447)
生物堿(Alkaloids)是一類含氮有機化合物,多數為含氮雜環結構,生物活性顯著,是中草藥中重要的有效成分之一[1]。如藜蘆堿是一類以西伐丁(Cevadine)、藜蘆定(Veratridine)為主的甾體類生物堿,具有降壓、抑菌及滅蟲作用,可添加在化妝品中用以除菌殺螨以及治療疥瘡皮炎等[2];麥角堿類可促進子宮和血管收縮,對神經系統有一定作用;秋水仙堿在臨床試驗中有抑制細胞增長的作用,可用于治療白血病、皮膚癌等。盡管生物堿大多具有顯著的生物活性,但同時也具有潛在的毒害性[3],如藜蘆堿是一種脂溶性神經毒素,可引起嘔吐、心律不齊、昏迷、呼吸抑制;麥角生物堿可引起失眠、幻覺及中樞神經效應[4];秋水仙堿在人體內產生的衍生物二秋水仙堿,會腐蝕人體腸胃和泌尿系統[5]。
近年來,隨著消費者對安全、健康、有效的護理方法的關注,越來越多的化妝品中開始添加純植物提取的中草藥成分,以達到提高保濕、抗菌、抗衰老等護膚功效。商家添加中草藥在追求功效的同時,有可能會引入生物活性顯著,但具有毒性的生物堿類化合物。長期使用這類化妝品將會給消費者的身體健康帶來損傷[6-7]。因此《化妝品安全技術規范》(2015版)[8]及歐盟化妝品法規(EC) No1223/2009[9]已明確將藜蘆堿及其鹽類、秋水仙堿及其鹽類、麥角菌屬產生的麥角堿類、蘿芙木引入的蘿芙堿等多種生物堿類列入禁用物質名單,但目前國內針對化妝品中這幾類生物堿的相關檢測方法及標準卻明顯滯后。因此,建立一個快速、高效、靈敏的化妝品中生物堿的檢測方法對于化妝品的安全性評價及風險監測均具有十分積極的意義。
目前生物堿的檢測方法主要集中在體液[10]、中草藥材[11]、農產品[12]及食品[13]等基質,所采用的分析方法有高效液相色譜法(HPLC)[14]、氣相色譜-質譜聯用法(GC-MS)[15]和液相色譜-串聯質譜法(LC-MS/MS)[16-18]等,其中,HPLC操作簡單,但檢測器的檢出限高,不適用于生物堿的痕量分析,且選擇性差,易出現假陽性;由于大多數生物堿具有熱不穩定性、高沸點,因此若采用GC-MS檢測方法,需對生物堿進行衍生化處理,操作復雜,重現性差;而LC-MS/MS法結合了高效液相色譜和質譜的特點,適用范圍廣、選擇性強、靈敏度高,特別是質譜的多反應監測技術可大大提高選擇離子的靈敏度,在痕量分析方面具有很大優勢,適用于化妝品這種基質復雜、目標物含量低的樣品的檢測分析。本文采用SPE凈化結合超高效液相色譜-串聯質譜法(UPLC-MS/MS)建立了一種同時測定化妝品中6種生物堿(秋水仙堿、蘿芙堿、麥角胺、藜蘆定、西伐丁、麥角克堿,結構式見圖1)的檢測方法,該方法操作簡單,定性定量分析準確,適用于化妝品中生物堿的安全性評價分析。
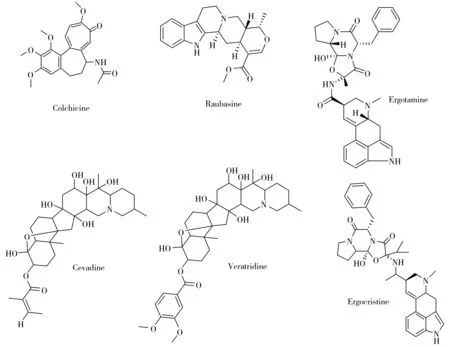
圖1 6種生物堿的分子結構式Fig.1 Molecule structures of six alkaloids
1 實驗部分
1.1 儀器與試劑
Thermo Ultimate 3000 超高效液相色譜儀及TSQ Endura三重四極桿質譜儀(美國Thermo公司);TurboVap LV氮吹儀(美國Caliper公司);KQ-250DV數控超聲波清洗器(昆山超聲儀器有限公司);3K15 離心機(德國Sigma公司);Milli-Q超純水器(美國Millipore公司);MS3渦旋儀(德國IKA公司)。
秋水仙堿、蘿芙堿購自德國Dr.Ehrenstorfer GmbH;麥角胺購自以色列Fermentek公司;麥角克堿購自加拿大TRC公司;藜蘆定、西伐丁鹽酸鹽購自美國IL公司;上述6種生物堿標準品的純度均大于97%。乙腈、甲酸、氨水、甲醇(色譜級)購自美國Merck公司;HLB固相萃取柱(500 mg/6 mL)購自CNW公司;Cleanert PCX-SPE(500 mg/6 mL)、Cleanert PEP(500 mg/6 mL)購自天津Agela Technologies公司。
樣品:隨機購買本地市售添加了植物草本的化妝品30種,其中包括10種爽膚水、10種膏霜類和10種香波類化妝品。取樣前混合均勻。
1.2 標準溶液的配制
生物堿混合標準中間液(100 μg/L):分別稱取適量的6種生物堿標準品,用乙腈配成單標儲備液(1 000 μg/L);然后分別吸取適量的單標儲備液,用乙腈稀釋成質量濃度均為100 μg/L的6種生物堿混合標準中間液。
生物堿混合標準工作液:吸取適量的生物堿混合標準中間液用空白基質提取液稀釋定容,得到質量濃度分別為0.2、0.5、1.0、2.0、5.0、10、20 μg/L的混合標準工作溶液。
所有標準溶液均保存于4 ℃條件下。
1.3 實驗方法
1.3.1樣品提取稱取0.5 g(精確至0.001 g)樣品于10 mL具塞比色管中,加入4 mL 0.1%甲酸水-乙腈(1∶3,體積比,下同),渦旋振蕩30 s使樣品混合均勻,超聲提取15 min,用0.1%甲酸水-乙腈(1∶3)定容至5.0 mL,渦旋混合均勻。吸取1.5 mL 提取液于高速離心管中,以10 000 r/min離心2 min,取1.0 mL上清液待凈化。
1.3.2樣品凈化依次用5 mL甲醇和5 mL水活化Cleanert PCX固相萃取柱,取1.0 mL上清液上樣,待溶液全部通過小柱后,用5 mL甲醇淋洗萃取柱,再用5 mL 5%(體積分數)氨化甲醇洗脫樣品,收集洗脫液,氮吹至近干,用0.1%甲酸水-乙腈(9∶1)定容至1 mL,渦旋混勻,過0.22 μm有機系濾膜后待測。
1.4 檢測條件
色譜柱:Thermo Hypersil GOLD 液相色譜柱(2.1 mm×100 mm×1.9 μm);柱溫30 ℃;進樣量為5 μL;流速0.3 mL/min;流動相:A為0.1%甲酸-乙腈,B為0.1%甲酸-水,梯度洗脫程序:0~3 min,60% B;3~6 min,60%~20% B;6~7 min,20%~60% B;7~8 min,60% B。
質譜條件:電噴霧離子源(ESI+);數據采集:多反應監測(MRM);離子化溫度:310 ℃,輔助氣流速:12 L/min;噴霧器溫度:330 ℃,鞘氣流速:40 L/min;電噴霧電壓:3 500 V;碰撞氣:氬氣,1.5 mTorr;化合物的母離子及定性定量離子、碰撞電壓和射頻電壓見表1。
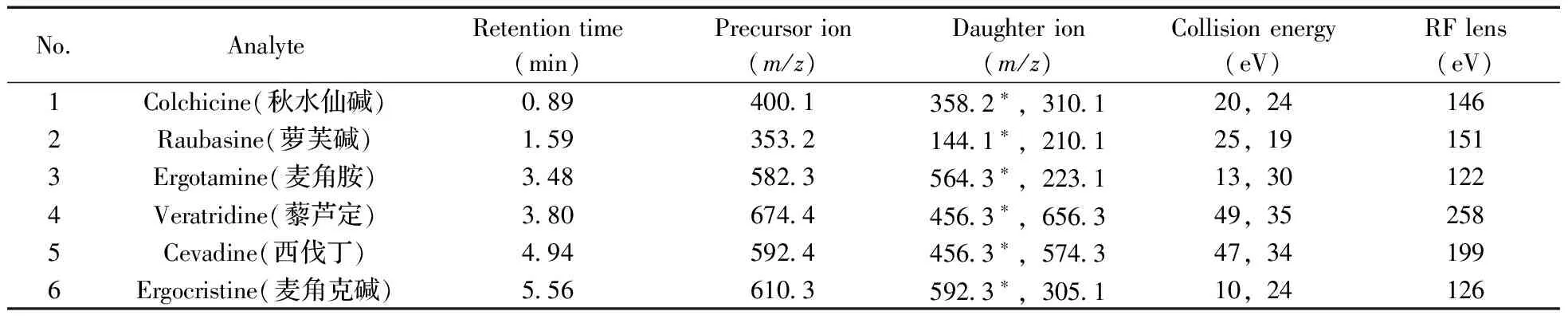
表1 6種生物堿的質譜參數Table 1 The multiple MS parameters of 6 alkaloids
*quantitative ion
2 結果與討論
2.1 檢測條件的優化
2.1.1色譜柱的選擇考察了Agilent Eclipse Plus C18(2.1 mm×100 mm×1.8 μm)、Waters ACQUITY UPLC BEH(2.1 mm×100 mm×1.7 μm)和Thermo Hypersil GOLD(2.1 mm×100 mm×1.9 μm) 3種液相色譜柱對6種生物堿的分離效果。結果發現:Agilent Eclipse Plus C18和Waters ACQUITY UPLC BEH柱對生物堿的保留能力較差,目標化合物峰形出現分叉,拖尾嚴重,而所有目標化合物在Thermo Hypersil GOLD柱上均能實現有效保留及分離,且重現性好,因此實驗選用該色譜柱對樣品進行分離。
2.1.2流動相的優化在正離子化模式下,水相中加入酸性介質有利于堿性化合物的正離子化,可以增強目標物的色譜峰響應。由于生物堿一般呈堿性,在流動相中加入乙酸銨可能會改善目標物的峰形和分離效果。因此實驗考察了流動相的水相分別為0.1%甲酸、0.1%三氟乙酸和10 mmol/L 乙酸銨時的分離效果。結果表明:0.1%甲酸作為水相時色譜峰響應最高,峰形對稱,分離度和靈敏度均能滿足檢測要求,可能因為呈堿性的生物堿與甲酸形成了中性的離子對化合物,與非極性固定相的作用增強,從而改善了分離效果,因此在水相中加入0.1%甲酸。分別以甲醇和乙腈作為有機相進行考察,結果顯示,兩種有機相下目標物的質譜響應相當,但有機相為乙腈時,色譜峰形更好、色譜柱柱壓更低,因此選用乙腈作為有機相。綜上,實驗選擇乙腈與0.1%甲酸混合溶液作流動相,此時目標物的檢測靈敏度高,分離度好而且能有效改善色譜峰的拖尾現象,可以滿足6種生物堿的檢測分析要求。
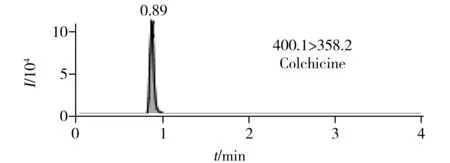
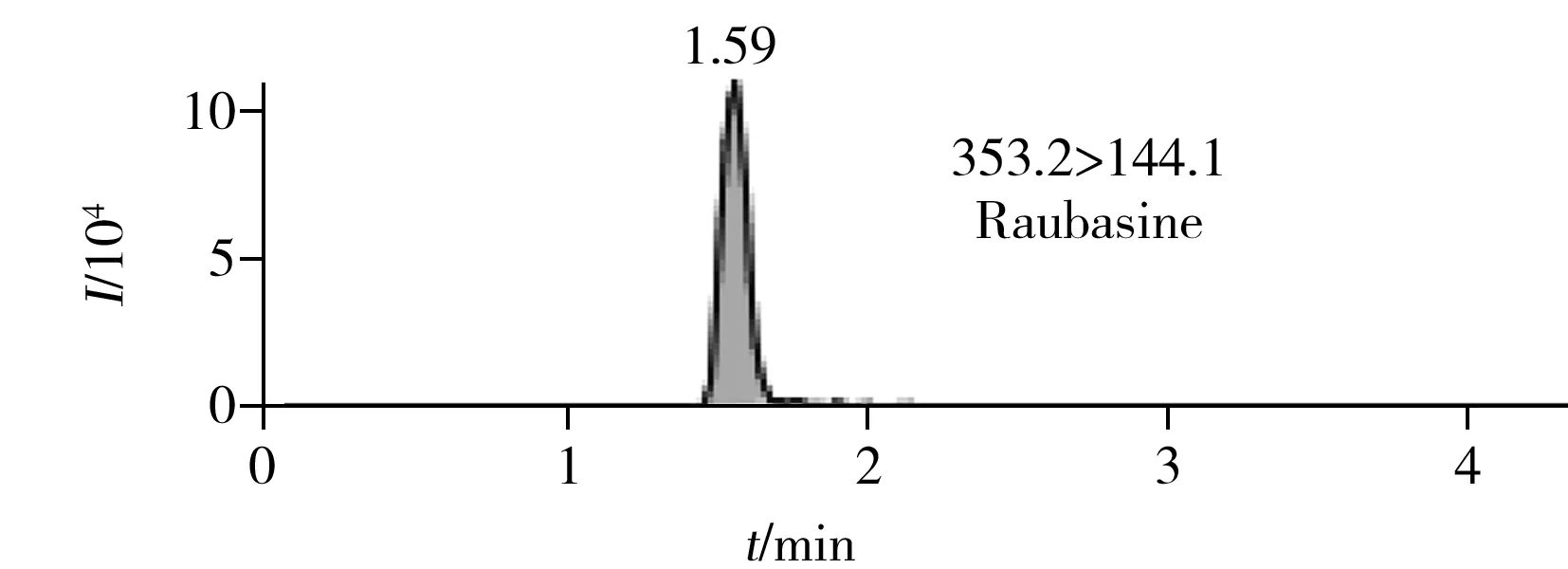
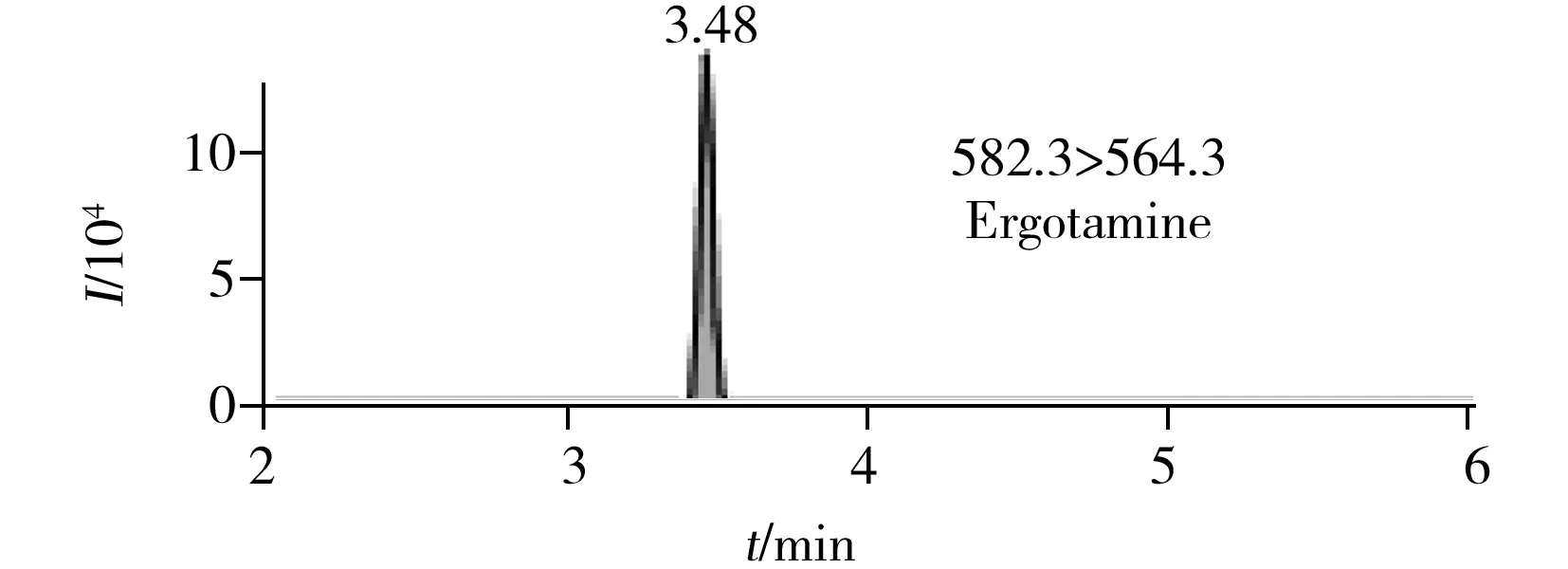
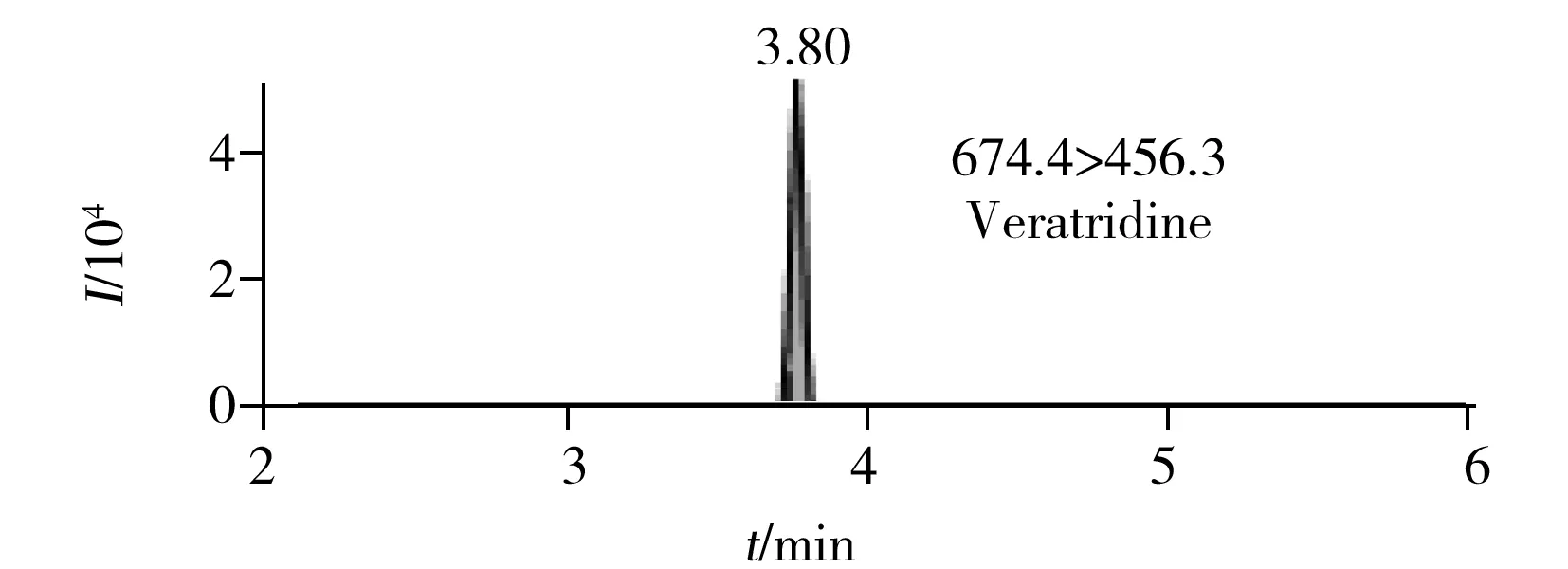
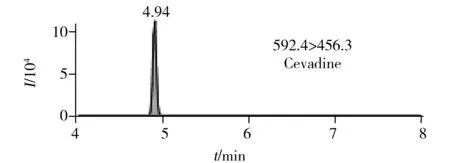
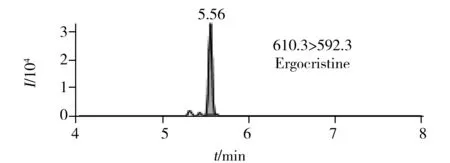
圖2 6種生物堿的提取定量離子色譜圖 Fig.2 MRM chromatograms of six alkaloids
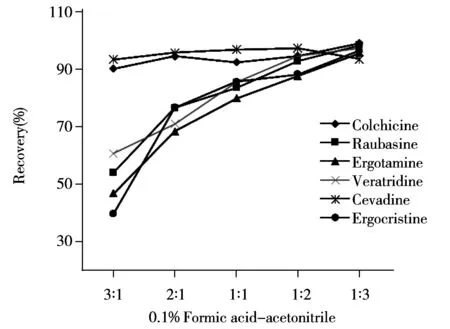
圖3 不同提取溶劑回收率的對比Fig.3 Recoveries comparison of different extraction solvents
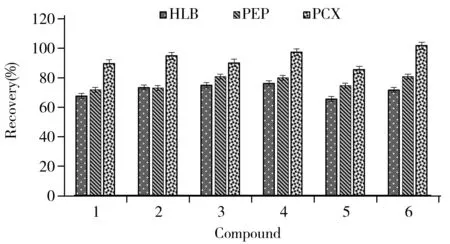
圖4 不同固相萃取柱回收率的對比Fig.4 Recoveries comparison of different SPE columns the compounds numbers denoted were the same as those in Table 1
在最優UPLC-MS/MS檢測條件下6種生物堿的提取定量離子色譜圖如圖2所示。
2.2 實驗條件的優化
2.2.1樣品提取方式的影響有文獻[19]對比了超聲水浴、渦旋振蕩和水平振蕩提取水、乳和霜類化妝品樣品的效果,由于膏霜類和香波類化妝品的黏度較大,超聲水浴提取可使樣品分散均勻,與提取溶劑接觸更充分。因此本實驗選用超聲水浴提取化妝品中的生物堿。由于提取時間的長短會對樣品的回收效果產生顯著影響,本實驗考察了不同超聲時間(0、5、10、15、20 min)對化妝品樣品中6種生物堿提取效率的影響。結果顯示,生物堿的回收率隨著超聲時間的延長而逐漸增大,到15 min之后回收率增長不明顯,為節約時間,本實驗的超聲時間選用15 min。
2.2.2樣品提取溶劑的影響化妝品的基質較為復雜,水溶性和脂溶性物質均較多,而6種生物堿的水溶性與脂溶性也存在差異,因此實驗選擇混合溶劑進行提取。生物堿的極性較大,且呈堿性,在酸性溶液中溶解性較好,所以采用0.1%甲酸-乙腈混合溶劑對膏霜類加標樣品進行提取。對比了不同配比的0.1%甲酸-乙腈溶劑對樣品的提取效果(如圖3)。結果表明,脂溶性好的蘿芙堿、麥角胺、藜蘆定和麥角克堿隨著乙腈比例的增加回收率逐漸增大;而秋水仙堿結構中存在酰胺基,西伐丁結構中存在酯基和羥基使得它們均具有較好的親水親脂性,因此經不同配比的0.1%甲醇-乙腈混合溶劑提取的回收率差異不大。實驗最終選擇以體積比為1∶3的0.1%甲酸-乙腈為提取溶劑,各待測物均能得到較好的回收效果。
2.2.3樣品凈化方法的影響生物堿類化合物作為禁用物質在化妝品中的含量通常很低,且化妝品中同時含有大量油脂和表面活性劑,基質復雜,若提取后直接上機,會對生物堿的檢測產生嚴重干擾,因此需對提取液凈化后再上機分析。目前報道的化妝品常用凈化方法有正己烷除脂[19]、PSA分散固相萃取凈化[20]及固相萃取柱凈化[21]等。固相萃取技術(SPE)是近年來發展迅速的一種樣品預處理技術,廣泛應用于化工、食品和環境等領域。它將液固萃取和柱液相色譜技術相結合,可將目標物與干擾物有效分離,回收效果好。因此本實驗選用SPE凈化提純目標物。由于固相萃取柱的種類較多,考慮到生物堿類化合物大部分呈堿性,極性較大,在硅膠和C18基質的固相萃取柱上保留較差,故選取以高分子聚合物為填料的Cleanert PEP 和Oasis HLB 以及適用于離子型化合物的Cleanert PCX柱進行研究。加標樣品提取液上樣后,Cleanert PEP和Oasis HLB先用5 mL水淋洗再用5 mL甲醇洗脫,Cleanert PCX用5 mL甲醇淋洗再用5 mL 5%氨化甲醇洗脫。所得回收率結果如圖4所示,3種SPE柱對6種生物堿均有一定的保留,Cleanert PCX的回收率最高,這可能是因為在酸性條件下生物堿類帶正電,凈化過程中首先通過正負電荷的相互作用緊密吸附在陽離子交換固相萃取柱上,然后用強堿將其置換下來,因此陽離子交換柱對6種生物堿的保留能力較強、選擇性好。本實驗選用Cleanert PCX對樣品進行凈化。
2.3 方法驗證
2.3.1基質效應化妝品基質較為復雜,基質引入的物質可能會干擾目標物的離子化效果從而影響其質譜響應。因此為消除樣品基質效應,本實驗采用空白基質提取液配制標準工作液,稱取適量空白樣品按照樣品前處理方法操作,制備空白基質溶液,將標準儲備液用空白基質溶液逐級稀釋得到系列標準工作溶液,使得標準工作溶液和樣品溶液均具有相似的離子化環境,從而確保了檢測方法定性與定量分析的準確性。
2.3.2線性范圍與檢出限在最優的色譜和質譜條件下,分別測定質量濃度為0.2~20 μg/L的系列6種生物堿的混合標準工作液,以混合標準工作液的質量濃度為橫坐標(x,μg/L),以各生物堿的定量離子對色譜峰面積為縱坐標(y)進行線性回歸計算,得到6種生物堿的線性方程(見表2)。結果表明,6種生物堿在一定質量濃度范圍內線性關系良好,相關系數均大于0.999 0。
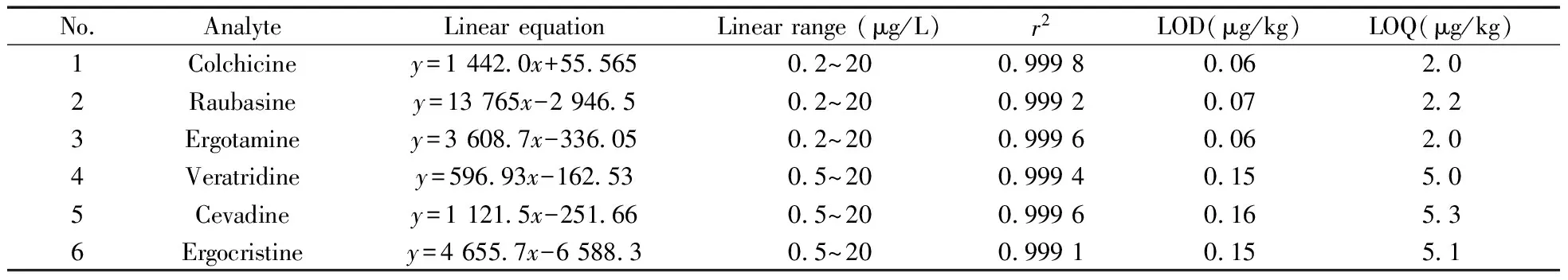
表2 6種生物堿的線性方程、線性范圍、相關系數、檢出限及定量下限Table 2 Linear equations,linear ranges,correlation coefficients(r2),LODs and LOQs of six alkaloids
通過測定質量濃度為0.2 μg/L(秋水仙堿、蘿芙堿、麥角胺)和0.5 μg/L(藜蘆定、西伐丁、麥角克堿)的標準工作液,計算得到各化合物的儀器檢出限(LOD,S/N=3)為0.06~0.16 μg/kg,結合前處理過程和回收率情況,最終計算得到6種生物堿的方法定量下限(LOQs,S/N=10)為2.0~5.3 μg/kg,結果見表2。該方法的靈敏度高,適用于化妝品中此6種生物堿的檢測分析。
2.3.3精密度與回收率由于化妝品的樣品基質各不相同,對方法的回收率及精密度影響較大。因而在最優實驗條件下,選取3種基質(爽膚水、面霜、香波)的陰性樣品進行加標回收實驗。每種基質分別添加3個濃度,每個濃度平行測定6次,計算回收率和相對標準偏差(RSD)。結果表明,3種加標濃度的回收率為83.9%~101.1%,RSD為0.7%~6.6%(見表3)。方法的回收率和精密度均能滿足日常檢測的要求。
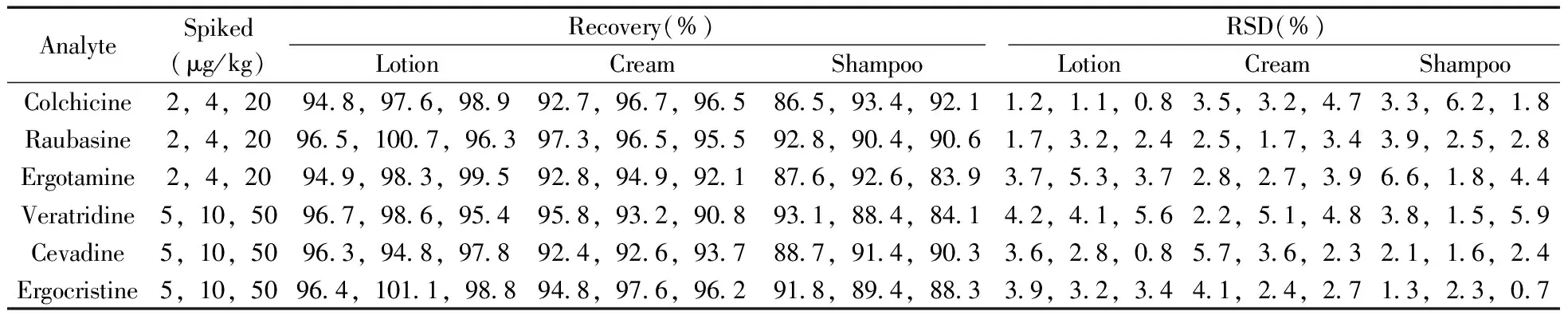
表3 6種生物堿的加標回收率及相對標準偏差(n=6)Table 3 Spiked recoveries and RSDs of six alkaloids(n=6)
2.4 實際樣品的測定
為評價該方法的有效性,本實驗測定了市售的爽膚水、膏霜、香波類等30種化妝品,所有樣品中6種生物堿的檢測結果均小于檢出限。
3 結 論
本文以0.1%甲酸水-乙腈混合溶劑超聲提取化妝品,Cleanert PCX柱凈化,經超高效液相色譜-串聯質譜法分離檢測后,可同時測定化妝品中6種生物堿。實驗通過對檢測條件、樣品前處理過程及凈化手段的優化,使得該方法應用于水、膏霜和香波3種化妝品基質中均能得到較好的回收效果及重現性。本方法操作簡單、可行性強、回收率高、結果準確、檢出限低,方法學各項技術指標均滿足定性、定量分析要求,適用于化妝品中有毒生物堿的測定,并為化妝品中生物堿禁用組分的風險監控提供了有效的技術支持。
參考文獻:
[1] Kittakoop P,Mahidol C,Ruchirawat S.Curr.Top.Med.Chem.,2014,14:239-252.
[2] Zheng S Z,Cui C Q,Liu Y Z.J.Med.Sci.YanbianUniv.(鄭善子,崔春權,劉永鎮.延邊大學醫學學報),2004,27(1):31-32.
[3] Cushnie T P,Cushnie B,Lamb A J.Int.J.Antimicrob.Agents,2014,44:377-386.
[4] Mavungu J D,Malysheva S V,Sanders M,Larionova D,Robbens J,Dubruel P,Peteghem C,Saeger S D.FoodChem.,2012,135:292-303.
[5] Song J P,Wang T,Chen X M,Yang J,Zhang F C,Shang J.Chin.Pharmacol.Bull.(宋金萍,王濤,陳雪梅,楊潔,張富春,尚靖.中國藥理學通報),2011,27(7):1019-1023.
[6] Gavaraskar K,Dhulap S,Hirwani R R.Fitoterapia,2015,106:22-35.
[7] Kozak M,Sobczak P,Zukiewicz-Sobczak W.HealthProblemsofCivilization,2016,10:64-70.
[8] People's Republic of China State Food and Drug Administration.Cosmetics Safety Specifications.2015 ed.Beijing:China Standard Press(中華人民共和國國家食品藥品監督管理總局.化妝品安全技術規范.2015版.北京:中國標準出版社),2016.
[9] European Parliament and the Council.Regulation(EC) No 1223/2009 of the European Parliament and the Council of 30 November 2009 on Cosmetic Products.[2009-12-22].http://eur-lex.europa.eu/legal-content/ EN/TXT/PDF/uri=CELEX:32009R1223&qid=1488848613844&from=en.
[10] Sun J N,Chen J,Shi Y P.J.Chromatogr.A,2014,1352:1-7.
[11] Su M X,Song M,Yang D S,Shi J F,Di B,Hang T J.J.Chromatogr.B,2015,990:31-38.
[12] Guo Q Z,Shao B,Du Z X,Zhang J.J.Agric.FoodChem.,2016,64:7033-7039.
[13] Ma X F,Liang T Z,Song W,Feng H B,Zhang Y.FoodSci.(馬曉斐,梁天佐,宋煒,馮浩彬,張巖.食品科學) ,2014,35(8):226-229.
[14] Qi L,Zhang J,Zhang Z Q.Chin.J.Chromatogr.(亓亮,張婧,張志琪.色譜),2013,31(3):249-253.
[15] Wu H Q,Zhang C H,Huang X L.J.Instrum.Anal.(吳惠勤,張春華,黃曉蘭.分析測試學報),2013,32(9):1031-1037.
[16] Lin A H,Su X C,She D,Qiu K,He Q.J.Chromatogr.B,2016,1008:65-73.
[17] Li C,Li J M,Gu L H.Pharm.Today(李純,栗建明,顧利紅.今日藥學),2018,28(3):152-154,161.
[18] Lorena L,Roberta M,Alessandra R,Clara M,Francesca C.FoodAnal.Methods,2016,9(6):1825-1836.
[19] Lin L,Zhang Y,Tu X K,Xie L Q,Yue Z F,Kang H N,Wu W D,Luo Y.Chin.J.Chromatogr.(林黎,張毅,涂小珂,謝麗淇,岳振峰,康海寧,吳衛東,羅耀.色譜),2015,33(3):275-281.
[20] Xun Z Q,Liu D H,Huang R R,He S,Hu D,Guo X D,Xian Y P.J.Sep.Sci.,2017,40(9):1966-1973.
[21] Luo H T,Huang X L,Wu H Q,Zhu Z X,Huang F,Lin X S,Ma Y F,Deng X,Zhou P C,Zhang Q Y,Jian Y T.J.Instrum.Anal.(羅輝泰,黃曉蘭,吳惠勤,朱志鑫,黃芳,林曉珊,馬葉芬,鄧欣,周培才,張秋炎,簡艷婷.分析測試學報),2016,35(2):119-126.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
