粉煤灰磁性吸附劑的制備及磷吸附機理
2018-08-01 01:54:56李建軍但宏兵IslamNazrul楊露敏葉先康朱金波
無機化學學報 2018年8期
李建軍 但宏兵 謝 蔚 Islam Nazrul 楊露敏 葉先康 朱金波
(1安徽理工大學材料科學與工程學院,淮南 232001)
(2伍倫貢大學超導與電子材料研究所,澳大利亞,伍倫貢 2500)
0 引 言
氮、磷等元素的含量超標是造成水體富營養化的主要原因[1]。研究表明,藻類等水生植物對氮、磷的需求比例符合經驗公式C106H263O110Nl6P[2],即氮、磷物質的量之比為16∶1。依據Leibig最小化法則(Leibig law of the minimum)可知,磷含量對水體富營養化的影響更為敏感[3],在治理水體富營養化方面,除磷比脫氮見效更快。現有的污水除磷方法包括化學沉淀、生物轉換及電解法等[4-5],這些方法雖具有一定的除磷效果,但仍存在除磷效果不穩定、污泥產生量大、適用性不廣等不足。在此背景下,吸附法成為頗具潛力的除磷方法。現有磷吸附劑包括有機高分子類和無機金屬氧化物及其鹽類。近年來,新型無機磷吸附劑以比表面積大、吸附速度快、性能與結構穩定等特點成為研究熱點[6-7],其中,含鑭化合物的磷吸附性能尤為突出。鑭系磷吸附劑對水中的含磷離子具有良好吸附能力,不但選擇性好,而且適用于低濃度含磷離子的吸附;其吸附磷后的產物對酸堿環境適應性好,不易產生二次污染;鑭離子的生物無毒性突出,有利于水資源的循環利用。鑭系磷吸附劑的研究主要集中在納米鑭化合物[8]、鑭化合物/非金屬(高分子)復合材料[9-10]、負載型鑭吸附劑等方面,其中,鑭負載膨潤土[11-12]研究最深入,部分產品已實現工業應用。鑭負載沸石/多孔材料[12-14]及鑭負載新型碳材料的研究方興未艾,促使磷吸附性能不斷提高[15-16]。為提高氧化鑭/氫氧化鑭的磷吸附性能,一些小組開始研究金屬離子共摻雜的(La,M)Ox的鑭系吸附劑[17]。
由于吸附法除磷要求吸附劑具有較大的比表面積并能較好的懸浮在水體中,因此磷吸附劑一般采用微細顆粒,由此造成吸附劑固液分離困難,此難題成為制約吸附法廣泛應用的瓶頸之一。近年來,一些研究者將磁分離技術引入污水處理中,以期利用磁場力促進吸附劑的固液分離[18]。這些研究多以化學合成的納米Fe3O4作為磁核[19-21]。由于納米Fe3O4合成工藝較復雜、生產成本和保存條件要求高且易造成二次污染,在一定程度上限制了磁性磷吸附劑的廣泛應用,因此尋找清潔廉價的磁核材料非常必要。粉煤灰磁珠來源于工業固廢粉煤灰,來源廣泛、價格低廉,由于其具有孔隙豐富、磁性強等特點,因而在重介質選煤、污水處理以及磁性復合材料制備等領域具有應用潛力[22-23]。如果將粉煤灰磁珠用作磁核處理則可望克服納米磁核的不足,為磁性磷吸附劑的工業化應用鋪平道路。同時,也可實現粉煤灰磁珠高附加值資源化利用,以廢治廢,環保效益和經濟效益顯著。粉煤灰磁珠作為磁核材料的主要障礙在于磁珠密度較大、顆粒較粗,因此在污水中懸浮性差、與水體作用面積不足。為此,需要對其進行預加工處理,以減小粒徑、增大比表面積。
本論文以精選、球磨處理后的粉煤灰磁珠作為磁核,通過化學沉淀法,制備了氧化鑭負載粉煤灰磁珠(CMS@La2O3)磷吸附劑。對所得樣品的形貌、結構以及磁性進行了系統表征,并將其用于吸附磷模擬污水中的含磷離子。在研究磷吸附影響因素及吸附動力學的基礎上,探討了磷吸附反應機理。
1 實驗部分
1.1 實驗藥品及儀器設備
實驗所用粉煤灰來源于大唐淮南洛河發電廠。化學試劑磷酸二氫鉀(KH2PO4),氯化鑭(LaCl3),四水合鉬酸銨((NH4)6Mo7O24·4H2O),抗壞血酸(C6H8O6),半水酒石酸銻鉀 (KSbC4H4O7·1/2H2O),氫氧化鈉(NaOH)均為分析純,購于上海國藥試劑有限公司。實驗用水為自制去離子水。
使用南京博蘊通有限公司的XGB04型行星式球磨機對粉煤灰磁珠進行球磨;采用煤炭科學研究總院生產的CXG-08SD型磁選管 (磁場強度0~350 mT連續可調)分選粉煤灰磁珠。采用日本島津XRD-6000型X射線衍射儀(XRD)分析樣品的晶體結構,掃描電壓40 kV,掃描電流30 mA,輻射源為Cu 靶 Kα(λ=0.154 nm),掃描角度范圍 10°~80°,掃描速度2°·min-1,并通過Jade6.0軟件與標準PDF卡片進行對比,確定樣品特征衍射峰。采用日本JEOL公司的JSM-7001F場發射掃描電子顯微鏡(SEM)和能譜儀(EDS),觀察樣品的形貌并進行能譜分析,工作電壓20 kV。利用美國康塔儀器有限公司的BET表面分析儀,通過氮氣吸附檢測樣品的比表面積和孔徑分布,脫氣溫度200℃。使用南京大學儀器廠生產的HH-20振動樣品磁強計(VSM)測量樣品的磁性。利用日本島津UV-2600型紫外可見分光光度計檢測含磷溶液的吸光度。
1.2 實驗方法
1.2.1 磁珠及磁性磷吸附劑的制備
磁珠預處理包括粉煤灰精細磁選和和球磨處理兩部分[24-25]。首先利用套篩對粉煤灰進行篩分,然后通過磁選管將大于100目的粉煤灰分別在300、100 mT磁場強度下進行多次磁選,獲得強磁性粉煤灰磁珠。利用行星式球磨機將所得粉煤灰磁珠在轉速為250 r·min-1的條件下球磨10 h,將所得磁珠微顆粒清洗并烘干備用。
通過化學沉淀法合成磁性磷吸附劑。取2.0 g磁珠微顆粒按固液比1∶100添加至200 mL 0.05 mol·L-1LaCl3溶液中,混合均勻,以 3 mL·min-1滴加1 mol·L-1NaOH溶液,調節溶液pH值至11,利用六聯電動攪拌器以500 r·min-1攪拌20 h。然后利用真空抽濾或磁分離分離混合液中的固體物,利用去離子水將固體物多次清洗至中性后,110℃真空干燥。最后將粉末樣品置入箱式電阻爐500℃下焙燒2 h,自然冷卻后經研磨、過篩備用。
1.2.2 磷吸附試驗
1.2.2.1 磷標準曲線的繪制
利用鉬酸銨分光光度法檢測磷含量。向7支50 mL具塞刻度管中分別加入質量濃度為2 mg·L-1的 KH2PO4溶液 0、0.5、1、3、5、10 和 15 mL, 使用去離子水稀釋至標線后,分別向其中加入10%(w/w)的抗壞血酸溶液1 mL,30 s后分別加入鉬酸鹽溶液2 mL,混合均勻后在室溫下靜置15 min。利用紫外可見分光光度計在波長為710 nm處檢測溶液吸光度并繪制標準曲線。
1.2.2.2 磷吸附試驗
稱取0.1 g磁性磷吸附劑添加至100 mL 20 mg·L-1KH2PO4溶液,調節溶液 pH=3,置于冷凍搖床25℃條件下,以160 r·min-1恒溫攪拌3 h,最后檢測上清液吸光度,并與標準曲線對比,計算磷的比吸附量。比吸附量的計算公式為:

式中,qt為比吸附量(mg·g-1),C0和 Ce分別為吸附前和吸附平衡時溶液中磷的濃度(mg·L-1),V為溶液體積(L),m為吸附劑的投加量(g)。
1.2.2.3 吸附劑再生
將吸附飽和的磁性磷吸附劑置于1 mol·L-1NaOH 溶液中,常溫下以 500 r·min-1解吸 3~6 h,磁分離其中的固體顆粒,洗滌至中性并充分干燥備用。再生吸附劑的磷吸附試驗步驟同(1.2.2.2)所述。
2 結果與討論
2.1 磁性磷吸附劑的結構與性能表征
2.1.1 磁性磷吸附劑的結構表征

圖1 (a)CMS、(b)磁珠微顆粒、(c)CMS@La2O3的SEM圖;圖(d)為圖(c)對應的EDS能譜;圖(e~h)為圖(c)對應的EDS面掃描圖像Fig.1 SEM images of(a)CMS,(b)CMS microparticles and(c)CMS@La2O3;(d)Corresponding EDS spectrum of CMS@La2O3in(c);(e~h)Corresponding EDS surface scanning images of CMS@La2O3in(c)

圖2 CMS和CMS@La2O3的(a)N2吸附-脫附曲線和(b)孔徑分布曲線Fig.2 (a)N2adsorption-desorption isotherm and(b)pore size distribution of CMS and CMS@La2O3
由樣品的SEM圖(圖1)可知,經球磨破碎后,磁珠顆粒的平均粒徑減小約1個數量級,形狀由類球形變成了不規則形狀。而經鑭改性后,磁珠顆粒表面有白色結晶物質生成,因此變得較為粗糙。EDS分析表明,改性后磁珠的能譜中出現了較強的鑭元素峰,鑭元素質量百分數可達28.76%。面掃描能譜圖表明,鑭改性樣品中除磁珠原有的O、Fe、Si、Al等元素外,廣泛分布有鑭元素,且在SEM圖中白色結晶物質對應位置上的鑭元素含量最高。說明磁珠表面的凸起包覆物應為鑭化合物,鑭元素較均勻地包覆在磁珠微顆粒表面。BET氮氣吸附結果顯示(圖2a),經氧化鑭包覆后,樣品的比孔隙率大幅增加,從磁珠微顆粒原有的2 cm·g-1增加到鑭包覆樣品的7 cm·g-1,孔徑分布圖(圖2b)表明氧化鑭包覆層具有豐富的孔隙結構,主要為孔徑低于6 nm的微介孔,這些孔隙結構對于提高樣品的物理吸附能力是有利的。
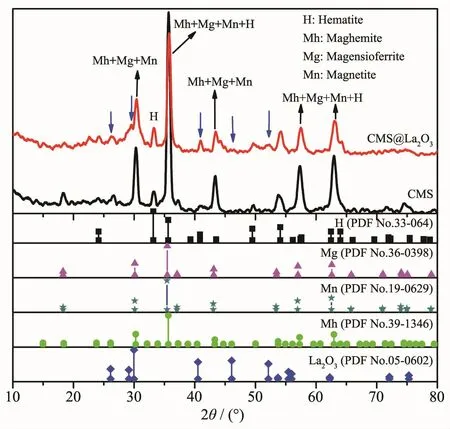
圖3 CMS和CMS@La2O3的XRD圖Fig.3 XRD patterns of CMS and CMS@La2O3
為確定樣品中鑭化合物的結構,對樣品進行了XRD分析。由圖3可知,原粉煤灰磁珠中含有磁鐵礦(magnetite)、鎂鐵礦(magnesioferrite)、赤鐵礦(hematite)和磁赤鐵礦(maghemite)等結構。CMS@La2O3的XRD圖中保留了原磁珠的所有衍射峰,說明改性過程未改變磁珠的結構。但多數衍射峰峰型有所寬化、強度有所下降,這是由于鑭化合物包裹在磁珠表面所致。值得注意的是,CMS@La2O3的XRD圖中在 27.18°、29.96°、40.54°、45.31°和 53.77°出現了 5個新的衍射峰,如圖中藍色箭頭所示,分別對應于氧化鑭(PDF No.05-0602)的(100)、(101)、(102)、(110)和(103)晶面。這說明磁珠微顆粒表面生成了氧化鑭晶體,與SEM及EDS表征結果相符。2.1.2 磁性吸附劑的磁性
圖4(a)是鑭改性前后磁珠的磁滯回線。由圖可知,磁珠微顆粒的比飽和磁化強度為39.80 emu·g-1,而CMS@La2O3的比飽和磁化強度為20.35 emu·g-1。這是由于經過氧化鑭負載后,樣品中磁性物質相對含量減少。雖然CMS@La2O3的磁性有所減弱,但仍具有較強鐵磁性,如圖4(b)所示,永磁體在距溶液2 cm外即可對CMS@La2O3實現高效磁分離。
2.2 磷吸附性能研究
2.2.1 磷標準曲線
由圖5可知,在波長為710 nm處測定磷的標準曲線:y=1.861 5x+0.005 7,其中,R2=0.999 6,這說明在質量濃度為0~0.6 mg·L-1的測量范圍內,定波長吸收強度與磷濃度具有較好的線性關系。
2.2.2 磷吸附量隨時間的變化
利用所得磁珠微顆粒和CMS@La2O3磁性吸附劑對P模擬污水進行吸附處理,試驗結果如圖6所示。磁珠微顆粒本身對含磷離子的吸附能力很弱,其飽和比吸附量只有2.07 mg·g-1;而經La2O3包覆后磷吸附能力顯著增強,比吸附量最高可達19.50 mg·g-1。從吸附曲線上看,CMS@La2O3對磷的比吸附量隨吸附時間的延長而逐漸增加,在磷吸附的初期,吸附速度很快,10 min內磷的去除率即可超過50%;當吸附時間超過60 min后,磷吸附速度迅速減緩,3 h后趨于飽和。與同類磁性磷吸附劑相比,CMS@La2O3吸附劑的磷比吸附量處于中等水平,具體吸附性能及吸附動力學模型對比如表1所示[9,13,18,26-31]。值得注意的是,該吸附劑在磷吸附過程中,溶液pH值隨著磷比吸附量增加的同時也在不斷增大。pH值增大說明在磷吸附過程中不斷產生氫氧根,意味著CMS@La2O3對含磷離子的吸附可能源于La2O3表面離子的配位不飽和引發的化學吸附[32]。La2O3表面與水分子配位-反應的過程如圖7所示,表面La離子首先與水分子配位,水分子的離解性化學吸著導致表面羥基化。羥基化的表面可與溶液中的磷酸根、磷酸氫根、磷酸二氫根等含磷離子發生離子交換,并釋放出氫氧根。其反應方程式如下:

圖4 (a)CMS和CMS@La2O3的磁滯回線;(b)CMS@La2O3顆粒被永磁體吸引分離Fig.4 (a)Magnetic hysteresis loops of CMS and CMS@La2O3;(b)CMS@La2O3particles are separated by the permanent magnet

圖5 磷的濃度標準曲線Fig.5 Standard curve of P concentration

圖6 吸附時間對CMS和CMS@La2O3磷吸附性能的影響Fig.6 Effect of adsorption time on the P adsorption properties of CMS and CMS@La2O3

表1 部分鑭系磷吸附劑的吸附性能及動力學模型對比Table 1 Comparison of adsorption performance and kinetics model of reported Lanthanide phosphorus adsorbents
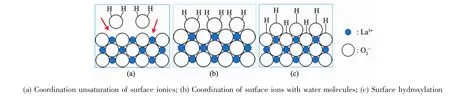
圖7 La2O3表面與水分子配位-反應的機理示意圖Fig.7 Schematic diagram of the coordination reaction between the surface of La2O3and water molecules
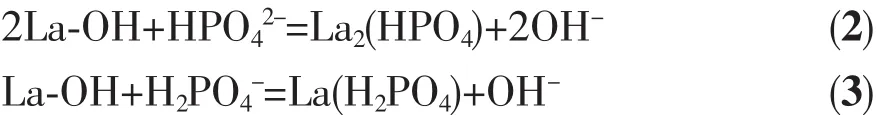
2.2.3 溶液pH值對磷吸附的影響
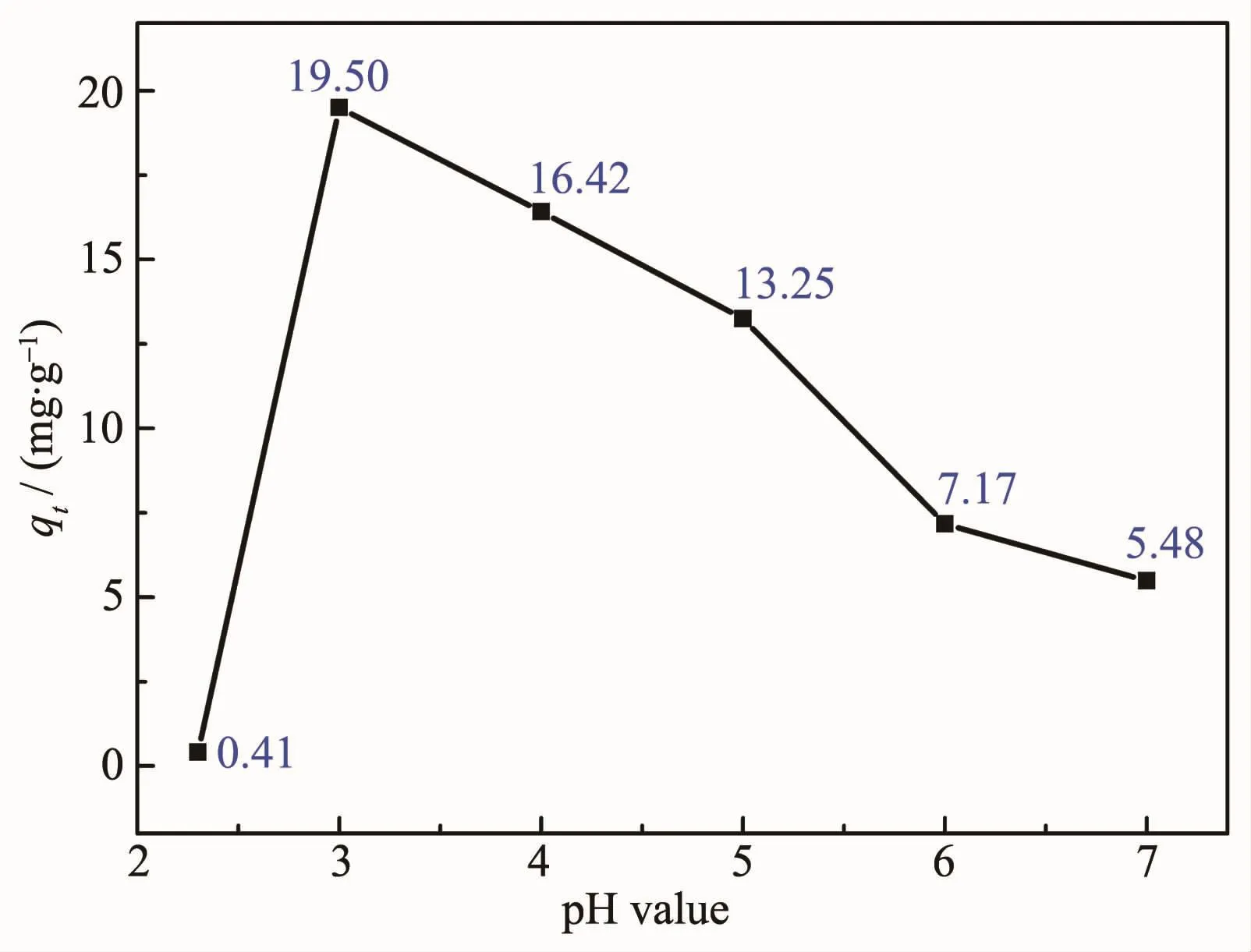
圖8 溶液pH值對CMS@La2O3磷吸附率的影響Fig.8 Effect of solution pH value on the P adsorption capacity of CMS@La2O3
圖8 為溶液初始pH值對磷吸附率的影響曲線。由圖可知,從中性水開始,磷的比吸附量隨pH值的降低而逐漸增加,當溶液pH值為3時,比吸附量達到最高;但當溶液pH值小于3時,磷吸附量迅速下降。含磷污水中的OH-會與含磷離子競爭吸附劑表面的活性位點[33],當溶液pH值不斷減小時,該競爭作用就會減弱,因而磷吸附不斷增強。同時,酸性增強會中和磷吸附反應中反應產物OH-,促進公式(2)、(3)反應的進行。此外,在不同pH值下磷酸鹽的種類也會發生變化,酸性環境下的磷酸鹽對氧化鑭的親和力較強[34-35],故在偏酸性的溶液中含磷離子的比吸附量要高于偏堿性的溶液環境。當溶液pH值低于3時,磷比吸附量迅速下降是因為此時溶液酸性過強,溶液中溶解平衡占據主導地位,鑭的羥基化合物趨于溶解[36-37],不利于反應(2)、(3)的進行,難以對磷酸根產生固定化作用[36]。此外,有研究表明,強酸性條件下不利于磷酸根吸附[33]。2.2.4 共存陰離子對磷吸附的影響
為探究溶液中常見陰離子對CMS@La2O3磷吸附的影響,分別做了不同濃度的氯離子、硫酸根和碳酸根干擾對比實驗。如圖9所示,不同陰離子的存在對CMS@La2O3的磷吸附效果的影響顯著不同。當KH2PO4污水中存在Cl-離子時,在 0~0.20 mol·L-1的濃度范圍內,磷的比吸附量波動范圍小于0.6 mg·g-1,說明Cl-離子對磷吸附干擾很小。而CO32-和SO42-離子對磷吸附的影響很顯著,當二者摻入離子濃度達到0.05 mol·L-1時,磷比吸附量分別下降8.38 和 6.61 mg·g-1。 這是由于 CO32-和 SO42-干擾離子與含磷離子之間對吸附位點產生了競爭吸附[38]。當干擾離子占據了氧化鑭表面的部分吸附位點,就會造成含磷離子吸附位點數不足、磷比吸附量下降。此外,CO32-在溶液中會水解釋放大量的氫氧根[39],氫氧根也會競爭吸附CMS@La2O3表面的活性位點。同時,La2(CO3)3的溶度積常數(3.98×10-34)低于LaPO4的溶度積常數(3.7×10-23)[40],說明 La2(CO3)3比LaPO4更難溶,故在溶液中,部分LaPO4會向La2(CO3)3轉化,從而釋放出部分磷酸根,使磷吸附量降低,故CO32-對CMS@La2O3磷吸附的抑制作用最強,與之前的研究結論相吻合[39-41]。與Cl-相比,當SO42-不斷積聚在CMS@La2O3吸附劑表面時,與磷酸根離子具有更強的靜電排斥作用[42],因此SO42-比Cl-具有更強的磷吸附干擾作用。
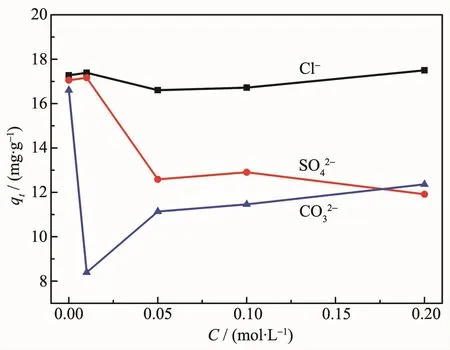
圖9 共存陰離子濃度(C)對CMS@La2O3磷吸附量的影響Fig.9 Effect of concentration of coexisting anions(C)on the P adsorption capacity of CMS@La2O3
2.3 吸附動力學研究
用于研究離子描述液-固吸附過程的動力學模型包括準一級與準二級動力學模型。其數學表達式為:
準一級動力學模型:

準二級動力學模型:

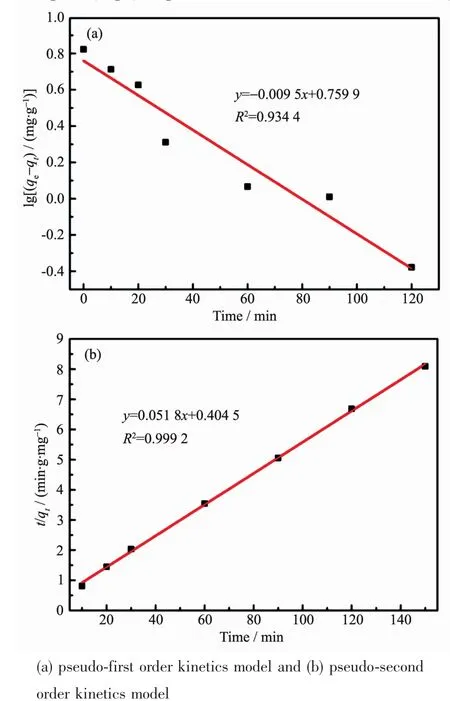
圖10 CMS@La2O3的動力學模型Fig.10 Kinetics model of CMS@La2O3
式中:qe與qt分別為吸附平衡時和吸附時間t時的比吸附量(mg·g-1),t為吸附時間(min),k1與 k2為吸附速率常數(min-1,g·mg-1·min-1)。
利用2種動力學模型對CMS@La2O3除磷過程進行擬合,如圖10所示。由圖可知,準一級方程的擬合相關系數R2為0.934 4,準二級方程的擬合相關系數R2為0.999 2。顯然準二級動力學模型的擬合相關系數R2更接近于1,而且由準二級方程計算得到的qe值(19.31 mg·g-1)也與實驗值(19.50 mg·g-1)更接近,以上結果表明準二級方程能更好地描述磷在氧化鑭負載粉煤灰磁珠表面的吸附行為,即CMS@La2O3吸附劑的磷吸附過程以化學吸附為主。由于離子交換作用屬于化學吸附過程,間接印證了前述公式(2)、(3)的磷吸附反應機理,說明CMS@La2O3的除磷機理為La2O3表面的羥基化-離子交換。
2.4 吸附劑的再生
吸附磷后的CMS@La2O3,可通過處理實現再生循環利用。具體方法可簡述為,將CMS@La2O3磁分離,置于 1 mol·L-1NaOH 溶液中,劇烈攪拌 3~6 h,然后清洗至中性,干燥后即可回用。如圖11所示,再生CMS@La2O3磷吸附劑前期的吸附速度依然很快,2 h后吸附趨于飽和,初次回收利用磷吸附劑的比吸附量為18.43 mg·g-1,可達原吸附劑的94.62%。隨著再生次數的增加,磁性吸附劑的磷比吸附量逐漸降低。
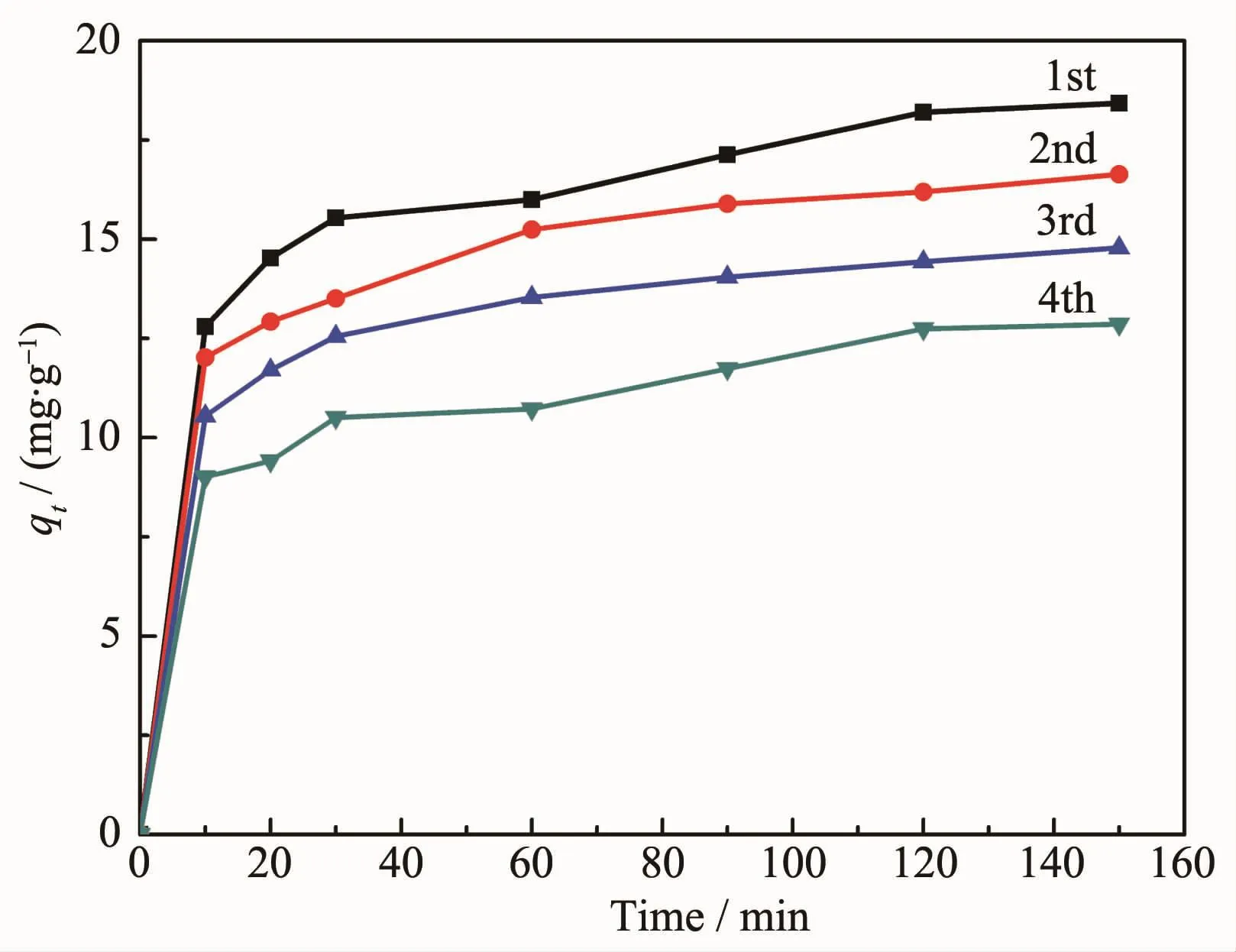
圖11 再生CMS@La2O3吸附劑的磷吸附曲線Fig.11 P adsorption curves of regenerated CMS@La2O3 adsorbent
3 結 論
(1)以粉煤灰磁珠微顆粒為磁核,通過化學沉淀法制備了CMS@La2O3磁性磷吸附劑,其比磁化強度可達20.35 emu·g-1,在外加磁場作用下,可實現高效磁分離。
(2)磷吸附試驗表明,當投加1 g·L-1CMS@La2O3磁性磷吸附劑處理pH值為3、濃度為20 mg·L-1的含磷污水時,其最高比吸附量為19.50 mg·g-1。CMS@La2O3吸附劑的磷吸附性能與吸附時間、溶液pH值以及共存陰離子有關。使用過的吸附劑經處理后可多次循環利用。
(3)CMS@La2O3的磷吸附過程符合吸附動力學準二級方程,以化學吸附為主。吸附反應機理為La2O3表面羥基化-離子交換。
