高糖環境中培養的小鼠心肌微血管內皮細胞蛋白激酶C活性、A激酶錨定蛋白150表達變化
2018-08-31 04:35:08曾超霍瑞雪鐘磊曾偉杰席玉勝范智文
山東醫藥 2018年31期
曾超,霍瑞雪,鐘磊,曾偉杰,席玉勝,范智文
(1中國人民解放軍第四二一醫院,廣州510318;2空軍軍醫大學唐都醫院)
近年來,糖尿病(DM)發病率逐年升高。我國有近一億DM患者,DM及其并發癥嚴重威脅著人類的健康[1]。冠心病是最嚴重的DM并發癥,已成為導致DM患者死亡的首要原因,但DM導致冠心病的具體發病機制仍不明確[2,3]。研究[4]發現,DM患者心肌微血管內皮損傷早于心肌細胞損傷,DM患者往往伴隨著心肌微血管內皮損傷等,而心肌微血管內皮損傷是冠心病發生、發展的基礎。蛋白激酶 C (PKC)參與了DM多種并發癥過程,尤其是心血管并發癥[5]。我們前期試驗發現,PKC的異常激活在DM心肌細胞損傷中扮演重要角色。A激酶錨定蛋白150(AKAP150)作為PKA、PKC、蛋白磷酸酶2B(PP2B)的錨定蛋白,在這些激酶的定位、分布及其對底物特異性作用中發揮著重要作用。AKAPs可通過調控PKC的活性狀態,促進DM患者的心肌細胞損傷,參與DM心血管并發癥的發生、發展[6]。但目前關于心肌微血管內皮細胞中PKC活性變化和AKAP150表達變化相關文章較少。2016年1~12月,我們觀察了高糖預處理小鼠心肌微血管內皮細胞PKC活性變化和AKAP150表達變化,并探討其意義。現將結果報告如下。
1 資料與方法
1.1 動物、儀器及試劑 健康雄性C57BL小鼠(8周齡,體質量18~22 g)10只,由空軍軍醫大學實驗動物中心提供。恒溫細胞孵箱(Thermo Scientific);超凈工作臺(蘇州精華設備廠);臺式離心機(Thermo);凝膠電泳及凝膠成像系統(Bio-Rad);掃描共聚焦顯微鏡(Olympus)。非放射性PKC活性試劑盒(Promega);AKAP150抗體(Millipore);PKC抗體(Millipore);β-actin抗體(Santa cruz);辣根過氧化物酶標記的二抗(北京中杉)。
1.2 心肌微血管內皮細胞的分離培養及高糖干預方法 取10只C57BL小鼠,處死后于無菌條件下取出左心室,置入平皿中,無菌 PBS沖洗左心室組織3次,75%酒精中泡洗15 s。去除左心室組織內外膜,用無菌眼科剪將組織剪碎為約 1 mm3的組織小塊。將組織塊放置于試管中,加入Ⅱ型膠原酶(濃度為0.2%)3 mL,37 ℃水浴消化6 min。加入3 mL胰酶(濃度為0.25%),37 ℃水浴消化5 min。加入3 mL含10%胎牛血清的DMEM培養液終止消化。100 μm濾網過濾,去濾過液,1000 r/min室溫離心10 min。棄上清,向消化后的細胞加入完全培養液(成份為:低糖DMEM培養液,15%胎牛血清,1%鏈霉素,100 U/L青霉素),均勻接種到培養瓶中,37 ℃、5% CO2孵箱中培養。細胞培養 6 h后換液,以去除未貼壁細胞,培養瓶中貼壁細胞為心肌微血管內皮細胞,24 h后再次換液,以后每 3 天換液一次,待細胞生長至80%融合后,消化傳代(0.25%胰蛋白酶)。取第 2~3代細胞,分為觀察組、對照組,分別加入含25、5 mmol/L葡萄糖的培養基。
1.3 心肌微血管內皮細胞PKC活性測定 細胞培養24 h時采用非放射性PKC活性試劑盒檢測細胞PKC活性。向DEAE 纖維素柱內加入1 mL PKC抽提液進行預平衡處理。待蛋白勻漿后,將蛋白勻漿液過柱。依次向0.2 mL 小離心管中加入實驗試劑,加入樣品或PKC,30 ℃ 再孵育30 min。把離心管放入沸水水浴或95 ℃ 加熱10 min終止反應。每個樣品中加1 μL 80%甘油。把10 μL樣品上樣到0.8%瓊脂糖凝膠孔中,100 V電泳15 min分開樣品,將瓊脂糖凝膠置于凝膠成像系統內測算磷酸化PKC與非磷酸化PKC比值,表示PKC的活性水平。參照PKC陽性對照,測量p-PKC、nonp-PKC的光密度(OD值),PKC活性水平= ODp-PKC/ODnonp-PKC。實驗重復3次,取平均值。
1.4 心肌微血管內皮細胞AKAP150蛋白檢測 采用Western blotting法。取對數生長期心肌微血管內皮細胞細胞1×107個,細胞裂解液常規提取細胞總蛋白,BCA法定量總蛋白,加入SDS上樣緩沖液于提取后的蛋白,煮沸5 min,取50 μg蛋白上樣,電泳,轉膜,使用 Bio-Rad 濕轉膜儀以恒流 300 mA轉膜 2 h,待蛋白轉至NC膜后,膜于3%BSA封閉液37 ℃封閉1 h,加入一抗 (anti-AKAP150,1∶1 000;anti-β-actin,1∶1 000) 中4 ℃孵育過夜,辣根過氧化物酶標記的二抗(1∶5 000),37 ℃孵育1 h。ECL發光液顯色,Bio-Rad凝膠成像系統成像Western blotting產物電泳條帶,Image-J軟件測量條帶灰度值。以條帶的灰度值與β-actin灰度值之比代表目的蛋白的相對表達量。實驗重復3次,取平均值。
1.5 心肌微血管內皮細胞PKC、AKAP150結合情況觀察 采用免疫熒光共定位法。取兩組原代心肌細胞,接種在1×1 cm2蓋玻片上,細胞按分組繼續培養后,以多聚甲醛(4 %)固定0.5 h,而后H2O2(3%)去離子水孵育5 min;山羊血清(10 %)封閉孵育10 min;加入AKAP150兔抗鼠和PKC鼠抗鼠(1∶200)抗體,4 ℃過夜,室溫下孵育2 h,復溫后,加入FITC標記的山羊抗小鼠(綠色)和羅丹明(紅色)標記的羊抗兔二抗,37 ℃孵育1 h;DAPI作用5 min,甘油密封;激光掃描共聚焦顯微鏡熒光激發,觀察細胞內的熒光分子的表達和共定位。FITC及羅丹明標記熒光強度升高提示PKC與AKAP150結合。
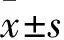
2 結果
觀察組、對照組細胞PKC活性分別為0.231±0.037、0.143±0.047,二者比較,P<0.05;觀察組、對照組細胞AKAP150蛋白相對表達量分別為0.527±0.080、0.124±0.038,二者比較,P<0.05。兩組細胞AKAP150蛋白電泳圖見圖1。

圖1 兩組細胞AKAP150蛋白電泳圖
對照組PKC與AKAP150結合較少,而觀察組FITC及羅丹明標記熒光強度升高,提示與對照組比較,觀察組細胞PKC與AKAP150結合明顯增多。
3 討論
DM是以慢性血糖升高為特征的代謝性疾病,因多種病因導致人體胰島素分泌相對或絕對不足,或者組織對胰島素敏感性下降。DM不僅僅是血糖升高,同時可導致脂類、蛋白質等多種物質代謝、轉化紊亂。長期、慢性的高血糖環境可以引起多種器官及系統并發癥,使DM并發癥發生率、致殘率、病死率明顯升高。近些年DM患者的發病率迅速上升,DM并發癥危害日趨嚴峻,以心血管并發癥尤為突出,約70%的DM患者最終死于心血管疾病[2]。DM的慢性高血糖環境導致心肌微血管損傷是DM心血管并發癥的重要原因之一,其機制包括心肌微血管內皮細胞連接不完整、屏障功能降低、心肌微血管密度降低、微血管數/心肌纖維數比率降低等,這些變化導致了心肌微循環障礙,造成心肌局部或整體氧供與血流量失衡,加重心肌缺氧易損性[7]。
近年PKC在心肌細胞及微血管內皮細胞中的作用受到關注[8]。PKC是絲氨酸/蘇氨酸激酶家族成員之一,根據蛋白結構、分布和底物結合上的差異,分為經典型PKC(cPKC)、非典型PKC(aPKC)和新型PKC (nPKC)[9]。PKC在全身組織細胞中均表達,作為細胞內重要信號傳遞分子,起到胞內信號傳遞的功能。普通情況下,PKC未被激活,定位在于胞質內,處于無活性狀態。如果接收上游分子信號后,PKC向細胞膜轉位,并被相關膜蛋白磷酸化后被激活。PKC的磷酸化狀態反映PKC激活水平。而PKC參與了DM心血管并發癥過程,在慢性高血糖狀態下,異常激活PKC在RAAS系統激活、心肌細胞氧化應激、鈣超載等損傷機制中起重要作用。相關報道[10]顯示,DM大鼠心肌組織中,均觀察到PKC在細胞膜、細胞核、細胞骨架和胞漿表達升高,且心肌細胞中總PKC活性增加。而在高糖預處理下,在新生乳鼠心肌細胞中觀察到PKCα表達增多且活性增加,通過激活下游通路NF-κB而致心肌細胞損傷。同時,在DM大鼠模型的心肌細胞中,PKC總活性增加,與大鼠心功能的下降相關[5]。而在DM豬模型中,心肌組織 PKC的轉錄水平明顯升高,并參與DM心血管損傷的過程[11]。我們觀察了DM條件下心肌微血管內皮細胞中PKC的活性狀態。結果顯示,與正常對照組相比,觀察組心肌微血管內皮細胞PKC活性明顯升高,這與上述研究中發現高糖預處理心肌細胞PKC活性升高一致。
AKAPs是激酶錨定蛋白家族成員之一,作為胞內錨定蛋白,介導了細胞外的信號向細胞內傳導,目前其生理功能及在多種疾病中的作用越來越受到關注。其具體作用是AKAPs錨定相關激酶結合后,將激酶轉移至在胞內合適部位被激活,并暴露激酶的活化位點,加快激酶對作用底物的快速反應,從而使底物更快、更容易被激酶活化而激活底物[12]。最初的研究發現AKAPs通過與PKA RⅡ亞基結合調節PKA的激活,加快信號的傳遞,且具有靶向特異性的特點。更進一步的研究發現生理狀態AKAPs在維持細胞生長、增殖,代謝,遞質傳遞等起到主要的作用,其定位于細胞膜、核膜、高爾基體、骨架復合體等,與多種激酶結合后,調節多種激酶的活性,從而調控信號通路的傳導。在心臟中,生理狀態下的AKAPs主要作用于心肌細胞的變時性、變力性及自律性等方面,而在病理狀態下,AKAPs在心力衰竭、心律失常、心肌重構、甚至心臟腫瘤等疾病中都起到相應作用[13]。
AKAP150是AKAPs家族主要成員,研究[14]發現激酶中有PKA、PKC、鈣調磷酸酶等能夠受AKAP150的調節,并通過調控激酶的活性作用于磷脂酶,環磷酸腺苷,鈣離子等信號通路的傳遞。我們前期研究發現,在心肌細胞中,AKAP150能夠與PKC錨定,加快PKC向細胞膜轉位及磷酸化水平,并加快PKC對下游信號分子的激活[6]。還有研究[15]報道AKAP150作用下PKC加快激活膜上離子通道,如L型鈣離子通道,鉀離子通道等。Navedo 等[16]敲除動脈血管平滑肌細胞的AKAP150分子,使用血管緊張素預處理后,觀察PKC轉位及激活基本消失,且未檢出鈣火花的發生,并提示PKC激活必要依靠AKAP150。而DM狀態下心肌微血管內皮細胞PKC的異常激活機制仍不明確。本研究結果顯示,與正常對照組比較,觀察組心肌微血管內皮細胞AKAP150蛋白相對表達量明顯升高,且與PKC活性增加相一致,免疫熒光共定位顯示高糖預處理小鼠心肌微血管內皮細胞AKAP150與PKC結合明顯增多,提示AKAP150可能在DM心肌微血管內皮細胞損傷中扮演重要角色,很有可能是通過調節PKC的活性而發揮作用的。我們下步工作是研究AKAP150在DM心肌微血管內皮細胞的具體損傷作用。但目前高血糖調節心肌微血管內皮細胞AKAP150表達水平的機制不明,本研究推測可能是高糖可作用AKAP150啟動子,從而增加AKAP150基因的轉錄,但需要進一步研究證實。
綜上所述,高糖預處理的小鼠心肌微血管內皮細胞PKC活性升高、AKAP150表達升高。PKC活性水平及AKAP150表達升高可能在高糖導致的小鼠心肌微血管內皮細胞損傷中起重要作用。
