不同溶劑中的苯乙烯-馬來酸酐共聚研究
2019-02-20 08:52:30鮑世軒郭雅妮
武漢工程大學學報 2019年1期
沈 榮,鮑世軒,郭 蓓,文 軒,郭雅妮
武漢工程大學材料科學與工程學院,湖北 武漢 430205
苯乙烯-馬來酸酐共聚物[poly(styrene-comaleic anhydride),PSMA]是由苯乙烯和馬來酸酐共聚而成。馬來酸酐由于空間位阻效應在一般條件下很難發生均聚,但和苯乙烯在靜電作用下極易形成一種電荷轉移絡合物,在引發劑作用下發生自由基聚合,形成典型的二元交替共聚結構[1]。其價格低廉,具有良好的耐熱性、耐磨性、裝飾性和尺寸穩定性;由于其分子骨架上含有極性親水基團馬來酸酐,PSMA還具有一定的生物降解性[2],廣泛應用于紙張施膠劑、黏合劑、乳化劑和顏料分散劑等方面[3]。近年來關于這種極具潛力的共聚物的研究非常活躍,包括PSMA的合成、化學改性、納米復合材料、共混改性和增韌改性等[4-6],其中對于PSMA共聚物的化學改性尤為引人關注。PSMA分子鏈含有酸酐及苯環單元,具有很強的反應活性及衍生能力,在較溫和的條件下易發生酯化、酰胺化、酰亞胺化,與堿發生酸堿中和反應,或使其帶上電荷等[7-13],也可對PSMA中的苯環進行磺化、硝基化、鹵化等改性,從而改變PSMA的親水性、親油性、柔性和熱穩定性等性能。通過分子設計和改造,合成出新的功能化聚合物,拓寬了PSMA的應用領域[14]。但這種改性會受PSMA中馬來酸酐含量高低的影響,所以合成具有高馬來酸酐含量的PSMA對其之后的改性會有很大幫助。
本文分別以乙酸乙酯、N,N-二甲基甲酰胺(N,N-Dimethylformamide,DMF)、甲苯為溶劑,選擇不同的原料摩爾比研究了在不同的溶劑中反應單體摩爾比對PSMA共聚物中馬來酸酐結構單元含量的影響。
1 實驗部分
1.1 實驗試劑
苯乙烯,馬來酸酐,偶氮二異丁腈(Azodiisobutyronitrile,AIBN),DMF,乙酸乙酯,甲苯。以上試劑均為化學純或分析純,其中苯乙烯經過減壓蒸餾處理,馬來酸酐利用氯仿重結晶,AIBN利用乙醇重結晶。
1.2 實驗方法
1.2.1 溶劑為乙酸乙酯 依次將1.2 mL(0.010 5 mol)苯乙烯,10 mL乙酸乙酯,1.03 g(0.010 5 mol)馬來酸酐,0.10 g AIBN加入單口燒瓶中。在N2氛圍下,75℃回流反應1.5 h,冷卻到室溫,以無水乙醇為沉淀劑使產物沉淀,抽濾得到白色粉末,在60℃烘箱中干燥12 h得到PSMA產物。改變原料中苯乙烯與馬來酸酐的配料摩爾比為1∶2和2∶1,其他條件不變進行反應。
1.2.2 溶劑為DMF 依次將5.7 mL(0.050 0 mol)苯乙烯,30 mL DMF,4.9 g(0.050 mol)馬來酸酐,0.164 g AIBN加入單口燒瓶中,N2氛圍下,60℃反應20 h后,冷卻到室溫,以無水乙醇為沉淀劑使共聚物沉淀,抽濾得到白色粉末,在60℃烘箱中干燥12 h。改變原料中苯乙烯和馬來酸酐的配料摩爾比為1∶2和2∶1,其他條件不變進行反應。
1.2.3 溶劑為甲苯 將5.7 mL苯乙烯和0.01 g AIBN溶解于20 mL甲苯中配成混合溶液,倒入恒壓漏斗中。將4.9 g馬來酸酐和100 mL甲苯加入單口燒瓶中,75℃下加熱攪拌0.5 h后,滴加苯乙烯和AIBN的混合溶液,滴加結束后75℃再恒溫反應0.5 h,將溫度升高到85℃再反應1 h,反應過程中出現的白色沉淀即為PSMA[15]。將反應物冷卻至室溫,抽濾,固體產物用甲苯洗滌2次,干燥得到產物PSMA。改變原料中苯乙烯與馬來酸酐的配料摩爾比為1∶2和2∶1,其他條件不變進行反應。
1.3 儀器與表征
采用傅里葉變換紅外光譜儀(美國Perkin Elmer公司,Spectrum Two型)(Fourier transform infrared spectroscopy,FT-IR)測 定 共 聚 產 物 的FT-IR圖,KBr壓片方法,掃描范圍為4 000~500 cm-1;采用酸堿滴定法對合成的PSMA中馬來酸酐的質量分數進行滴定測試,配置0.1 mol/L的HCl和NaOH溶液,稱取0.15 g PSMA于50℃溶于30 mL的0.1 mol/L的NaOH溶液中,自然冷卻至室溫后,滴加幾滴甲基紅溶液于其中,再用0.1 mol/L的HCl溶液滴定至溶液變紅。記錄下消耗HCl溶液的體積VHCl,再根據如下公式可以得到PSMA中馬來酸酐的質量分數:

采用核磁共振波譜儀(400MR型,Agilent公司)測定PSMA的核磁共振氫譜(1H nuclear mag?netic resonance spectroscopy,1H-NMR),溶劑為二甲基亞砜;采用凝膠滲透色譜(RID 20型,Shimadzu公司)(gel permeation chromatography,GPC)儀測定PSMA的相對分子質量,溶劑為四氫呋喃;采用綜合熱分析儀(STA 409 PC型,NETZSCH公司)測定PSMA 的 熱 重 分 析(thermogravimetric analysis,TGA)曲線,溫度范圍為40~800℃,升溫速率為10℃/min,氮氣氛圍。除了馬來酸酐質量分數的測定外,其余測試對象均為以甲苯為溶劑、n(苯乙烯)∶n(馬來酸酐)=1∶1時合成的產物。
2 結果與討論
2.1 合成條件對共聚組成比的影響
表1為不同溶劑和原料摩爾比條件下合成得到的PSMA產物,通過酸堿滴定法測定的共聚物中的馬來酸酐質量分數及聚合物的產率。由表1可知,以乙酸乙酯和DMF為溶劑進行均相溶液聚合時,3種單體摩爾比下,產率均大于90%,但聚合物中馬來酸酐結構單元的含量均低于40%,即聚合物中可能含有大量的無規共聚物結構和苯乙烯均聚物;以甲苯為溶劑進行沉淀聚合的3種不同的單體摩爾比中,當苯乙烯與馬來酸酐的原料摩爾比為1∶1時,合成的PSMA中馬來酸酐的質量分數最高(酸堿滴定法測得為40.26%),產率為75%。甲苯中的3組聚合反應(實驗編號7-9)產率低于前6組的均相溶液聚合的產率,是因為產物中的苯乙烯均聚物已溶于甲苯中。

表1 在不同溶劑及原料摩爾比下聚合物中馬來酸酐的質量分數Tab.1 Mass fraction of maleic anhydride in polymer with different solvents and material mole ratios
2.2 結構表征
2.2.1 FT-IR表征 圖1(a)為編號7條件下合成的PSMA的FT-IR圖:1 458 cm-1處、1 507 cm-1處和1 632 cm-1處為苯環上碳碳鍵面內伸縮振動吸收峰;767 cm-1處和701 cm-1處的特征峰為苯環中碳氫鍵的面外彎曲振動峰;1 083 cm-1處為苯環上碳氫鍵的面內彎曲振動特征峰;3 021 cm-1處為苯環上碳氫鍵伸縮振動吸收峰;1 740 cm-1為羰基的伸縮振動吸收峰:2 922 cm-1處為飽和碳氫鍵的特征峰;1 776 cm-1處和1 857 cm-1處的吸收峰分別為馬來酸酐中碳氧雙鍵的對稱和反對稱伸縮振動的吸收峰,1 220 cm-1處的吸收峰為酸酐基團中碳氧單鍵的伸縮振動吸收峰。因此可以證明所合成聚合物為苯乙烯-馬來酸酐共聚物[16]。
2.2.21H-NMR表征 圖1(b)為編號7條件下合成PSMA的1H-NMR圖,其中7左右的峰為聚合物中苯環的特征峰,而2~4的峰為甲基和亞甲基的特征峰,對1H-NMR譜圖中6.0~7.7及2.2~3.8的峰面積進行積分,再根據公式

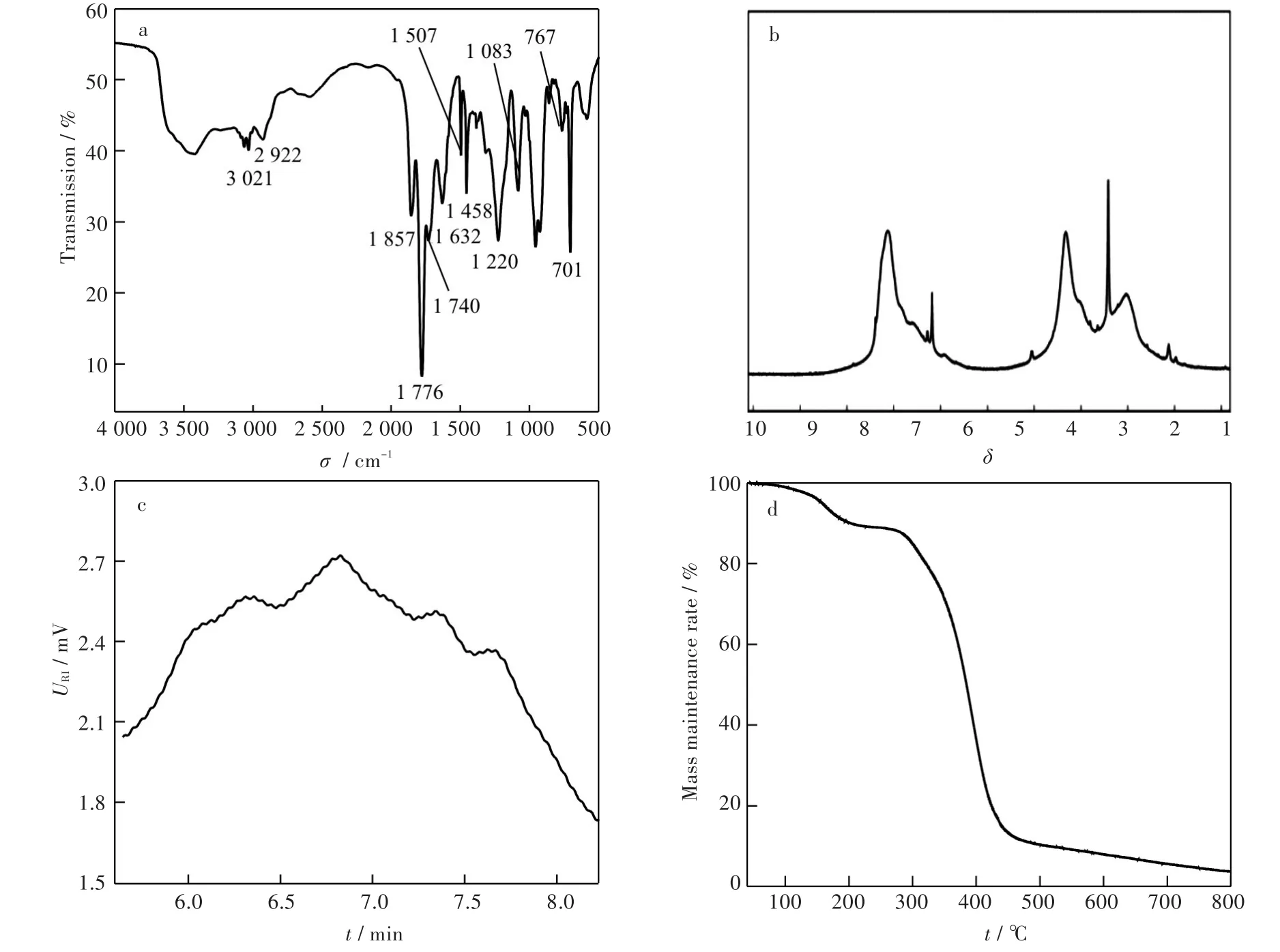
圖1 PSMA的表征圖譜:(a)FT-IR譜圖,(b)1H-NMR譜圖,(c)GPC圖譜,(d)TGA曲線Fig.1 Characterization spectra of PSMA:(a)FT-IR spectrum,(b)1H-NMR spectrum,(c)GPC chromatogram,(d)TGA curve
可以計算得出PSMA中馬來酸酐的摩爾分數[17],由此可算得PSMA中馬來酸酐的質量分數為46.42%,接近于PSMA交替共聚物中馬來酸酐的質量分數(48.49%)。
2.2.3 聚合物分子量分析 圖1(c)為編號7條件下合成的PSMA的GPC圖譜,重均分子量和數均分子量分別為79 697和35 185,分子量分布指數為2.27,說明聚合物的相對分子量分布比較集中。
2.3 聚合物熱失重分析
圖1(d)為PSMA的TGA曲線:100~230℃之間的失重來源于PSMA中酸酐基團的熱分解,而300~450℃之間的失重為共聚物主鏈受熱斷裂進而分解導致的失重。
3 結 語
以AIBN為引發劑,分別以乙酸乙酯、DMF、甲苯為溶劑,通過改變苯乙烯與馬來酸酐的投料摩爾比,探究了PSMA的合成條件對共聚物中馬來酸酐單元含量的影響。當以甲苯為溶劑進行沉淀聚合反應,苯乙烯和馬來酸酐的投料摩爾比為1∶1時,PSMA的產率為75%,通過1H-NMR測得的PSMA共聚物中馬來酸酐單元的質量分數為46.42%,接近于交替共聚物中馬來酸酐質量分數;通過GPC測得PSMA產物的重均分子量為79 697。本實驗所述方法具有合成工藝路線簡潔,后處理操作步驟簡便,產率高等優點;所制備的產品是一種低聚合度、高馬來酸酐單元含量的共聚物。
