宜昌綠肉獼猴桃果肉中葉綠素的結構鑒定及其抗氧化活性
2019-02-20 02:55:26
食品工業科技 2019年23期
關鍵詞:質量
(湖北省生物酵素工程技術研究中心,三峽大學食品生物技術研究中心,生物與制藥學院,湖北宜昌 443002)
作為天然色素的葉綠素具有抗氧化能力,可應用于醫療、食品、化妝品等行業[1-2],近年來對植物葉片和蔬菜中葉綠素的抗氧化性研究日益增多,但對水果中的葉綠素抗氧化性研究仍然較少。研究發現,葉綠素及其衍生物都具有一定的抗氧化能力,且獼猴桃果肉中的已知的葉綠素主要有葉綠素a、葉綠素b、脫鎂葉綠素a、脫鎂葉綠素b、植基葉綠素a、植基葉綠素b、脫鎂葉綠酸鹽a[3],但其中葉綠素的種類和含量因原料的不同差異也較大[4],因此本實驗在鑒定獼猴桃中葉綠素及其衍生物結構基礎上,進行抗氧化性研究。其中葉綠素母系結構以葉綠素a和葉綠素b為例,分子量分別為893和907,化學結構式如圖1所示[5]。

圖1 葉綠素a,b的化學結構Fig.1 Chemical structure of chlorophyll a and b
DPPH·是一種穩定的自由基,在有機溶劑中呈紫色,在517 nm波長下有較大吸收,當加入抗氧化劑后一部分自由基被清除,使在該波長下吸收降低,以此來評估物質的抗氧化活性[6];Fe3+在測定化合物提供電子的情況下可被還原為Fe2+,吸光值越大還原能力越強,即物質的抗氧化性越好[7]。馬惠與程茵等[8-9]都采用DPPH法與鐵離子還原法進行了抗氧化性研究;馬可純[10]在研究獼猴桃復合粉抗氧化研究中,以DPPH·清除能力和還原能力結果為指標,發現華優鮮果的DPPH·清除能力和還原能力最高。
據近幾年研究表明,葉綠素及其衍生物具有一定的抗氧化活性,獼猴桃果實除高含量VC和多酚等抗氧化成分外,張麗華等[11]研究了提取液的抗氧化活性,但對其內葉綠素及其衍生物的純品沒有研究。本研究以宜昌綠肉獼猴桃為原料,通過柱層析、質譜和核磁等技術,對果肉中葉綠素進行組分分析與結構鑒定,得到葉綠素單品和其降解產物,為研究獼猴桃在加工過程中的變色機理提供理論支撐,通過DPPH法和Fe3+還原力法對其單品進行了抗氧化活性研究,為今后研究其穩定性、藥理性質等奠定物質基礎。
1 材料與方法
1.1 材料與儀器
新鮮綠肉獼猴桃 從當地市場購買;甲醇、石油醚、三氯化鐵、DPPH(2,2-二苯基-1-1苦肼基)二氯甲烷、無水乙醇、乙酸乙酯、丙酮(均為分析純),甲醇、乙腈(均色譜純)、氘代甲醇和氘代氯仿 天津市天力化學試劑有限公司;硅膠Gf254薄層色譜板 青島海洋化工廠;中性氧化鋁(100~200目)、D101大孔樹脂 天津市恒興化學試劑制造有限公司。
EV312旋轉蒸發器 北京萊伯泰科儀器股份有限公司;UV-1800紫外可見分光光度計 島津制作所;6210 ESI/TOF質譜儀 美國安捷倫;1200系列液相色譜儀 美國安捷倫;Bruker AVANCE III HD 400 MHz超導核磁共振譜儀 美國布魯克·道爾頓公司(Bruker Daltonics Inc.)。
1.2 實驗方法
1.2.1 獼猴桃葉綠素的制備 獼猴桃去皮打漿后,用丙酮∶甲醇體積比1∶1,液料比1.22 mL/g、提取時間236 min、提取溫度59 ℃提取,提取液于4000 r/min離心10 min,分離出上層提取液(黃綠色),剩余果渣用相同的提取方法再次提取,合并兩次提取液,減壓濃縮后標記為脂溶性色素待測,冰箱4 ℃下密閉避光保存。剩余果渣用蒸餾水與無水乙醇體積比1∶4,液料比1.22 mL/g、提取時間236 min、提取溫度59 ℃提取,提取液于4000 r/min離心10 min,分離上層提取液(黃褐色),減壓濃縮后標記為水溶性色素待測[12],于冰箱4 ℃下密閉避光保存。
1.2.2 柱層析對獼猴桃色素的分離純化
1.2.2.1 填料預處理 中性氧化鋁:稱取250 g,加石油醚攪拌,浸泡10 min[13];D101大孔樹脂[14]:稱取100 g樹脂,95%乙醇浸泡24 h,充分溶脹后,用95%乙醇淋洗至流出液與水混合(1∶3)不呈白色渾濁,之后以蒸餾水洗凈乙醇。
1.2.2.2 洗脫條件 用D101樹脂柱分離純化水溶性色素,各濃度均用1000 mL乙醇進行洗脫,濃度分別為20%、40%、95%,流速為1.5 mL/min,樹脂分離純化時上樣量為10 mL,分離得到的不同餾分用Gf254薄層色譜板點板,展開劑為丙酮∶二氯甲烷∶甲醇3∶6∶1;用氧化鋁柱分離純化脂溶性色素,用1000 mL石油醚:丙酮混合液進行洗脫,比例分別為100%石油醚、10%、25%、50%、100%甲醇,柱層析分離純化時上樣量為15 mL,分離得到的不同餾分用Gf254薄層色譜板點板,展開劑為石油醚∶丙酮∶甲醇 8∶1∶1。
1.2.3 質譜檢測 在JASCO J-815CD光譜儀上,采用0.1 cm和1 cm的石英試管,在室溫下,采用100 nm/min掃描速率、2 nm分辨率和32次掃描,用干燥氮氣沖洗,獲得ECD光譜。樣品在甲醇中以10-4mol/L或10-3mol/L的濃度溶解,并用溶劑減去光譜。
1.2.4 核磁檢測 樣品在Bruker AVANCE III HD 400 MHz超導核磁共振譜儀上進行測試,溶劑為氘代甲醇和氘代氯仿,1H譜的工作頻率為400 MHz,脈沖序列為zg30,空掃次數(DS)為2次,采樣時間(AQ)為3.985 s,采樣次數(NS)為16次,增益值(RG)為101;13C譜的工作頻率為100 MHz,脈沖序列為zgpg30,空掃次數(DS)為4次,采樣時間(AQ)為1.363 s,采樣次數(NS)為1580次,增益值(RG)為161。
1.2.5 抗氧化活性研究
1.2.5.1 提取物對DPPH·清除能力的測定準確稱取冷凍干燥后的各個待測樣品及VC樣品50 mg,分別用溶劑溶解并定容至25 mL,配制成質量濃度為2 mg/mL溶液保存備用。
將備用溶液用蒸餾水稀釋至所需的各個濃度,取3 mL各濃度樣品溶液加入3 mL 0.16 mmol/L DPPH乙醇溶液于25 ℃水浴加熱15 min后,在517 nm測溶液吸光度(A1),每個濃度做3個平行實驗,取平均值,同時以3 mL樣品中加入3 mL蒸餾水測吸光度(A2),取3 mL蒸餾水代替樣品加入3 mL DPPH溶液測吸光度(A0)。以VC為對照,按下列公式計算DPPH·清除率,并計算自由基清除率IC50值[12-13]。
1.2.5.2 提取物對Fe3+還原力的測定 準確樣品100 mg,溶劑溶解并定容至25 mL,配制質量濃度為4 mg/mL樣品溶液,稀釋成所需的不同質量濃度,取2.5 mL各濃度的樣品溶液,加入2.5 mL 0.2 mol/L磷酸緩沖液和2.5 mL 1%鐵氰化鉀,50 ℃水浴20 min后急速冷卻,加入2.5 mL 10%三氯乙酸溶液混勻,0.2 μm水相濾膜過濾,取上清液5 mL,加入5 mL 0.02%三氯化鐵溶液混勻,10 min后在700 nm波長下測定吸光度。吸光度越大則還原能力越大,以VC作為陽性對照[14]。
1.3 數據處理
所有試驗均重復3次,取平均值,實驗數據采用Microcal Origin 6作圖,IC50使用IC50計算器計算。
2 結果與分析
2.1 柱層析分析
通過中性氧化鋁柱層析分離純化,得到4個脂溶性色素餾分,分別為組分A1100%石油醚餾分(黃綠色)、組分B1石油醚∶甲醇10∶1餾分(橄欖綠)、組分C1石油醚∶甲醇4∶1(藍綠色)、組分D1100% 甲醇餾分(黃綠色);通過D101樹脂柱分離純化,得到2個水溶性色素餾分,分別為組分E120%乙醇餾分(黃褐色)、組分F140%乙醇餾分(黃褐色)。其中組分C1根據文獻可知為葉綠素a[15]。
2.2 質譜檢測分析
圖2是以組分A1、D1為例,ESI+-Q-TOF-MS測得[M+H]+的準確質量及其主要特征碎片離子m/z的圖譜。最終得到物質A1的[M+H]+的準確分子質量為927.6967,與分子式C55H84O7N4相應;物質B1的[M+H]+的準確分子質量為588.4304,與分子式C34H33O5N4相應;物質D1的[M+H]+的準確分子質量為601.1116,與分子式C35H34O5N4相應;物質E1的[M+H]+的準確分子質量為927.6985,與分子式C55H84O7N4相應;物質F1的[M+H]+的準確分子質量為701.5039,與分子式C37H38O9N4相應。
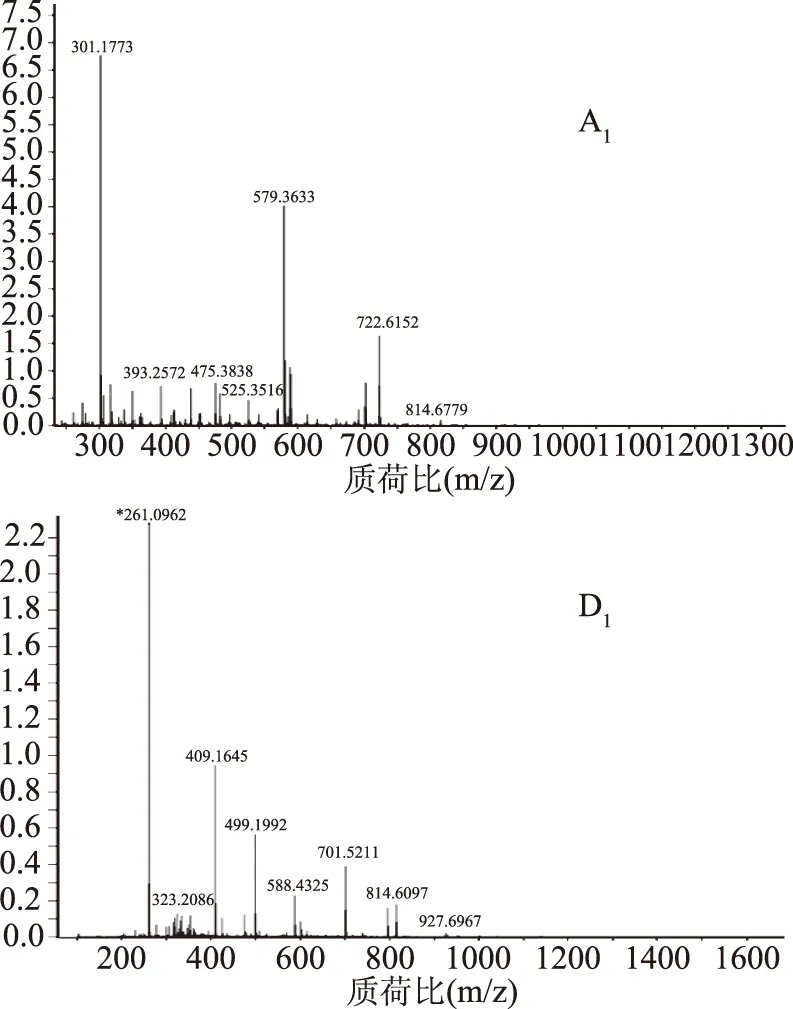
圖2 組分A1,D1的物質母離子的準確質量和離子組成圖譜Fig.2 Accurate mass and ion composition atlas of material mother ions of components A1 and D1
2.3 核磁檢測分析
圖3是以A1、D1為例的1H和13C NMR譜。在13C NMR譜中,A1的δ 22.73、27.23、29.70、31.96、62.13、68.89、127.77、128.26、130.25;B1的δ 22.87、27.38、29.75、65.24、127.93、128.43、130.42、132.15、174.06;D1的δ22.85、29.49、37.93、126.84、127.38、129.75、179.07;E1的δ12.92、62.81、64.51、68.01、69.86、70.48、74.90、75.39、76.70、97.82;F1的δ61.37、61.47、64.50、68.00、70.48、74.90、76.70、97.82,與文獻[16-19]中香蕉的葉綠素及降解產物核磁數據高度吻合。
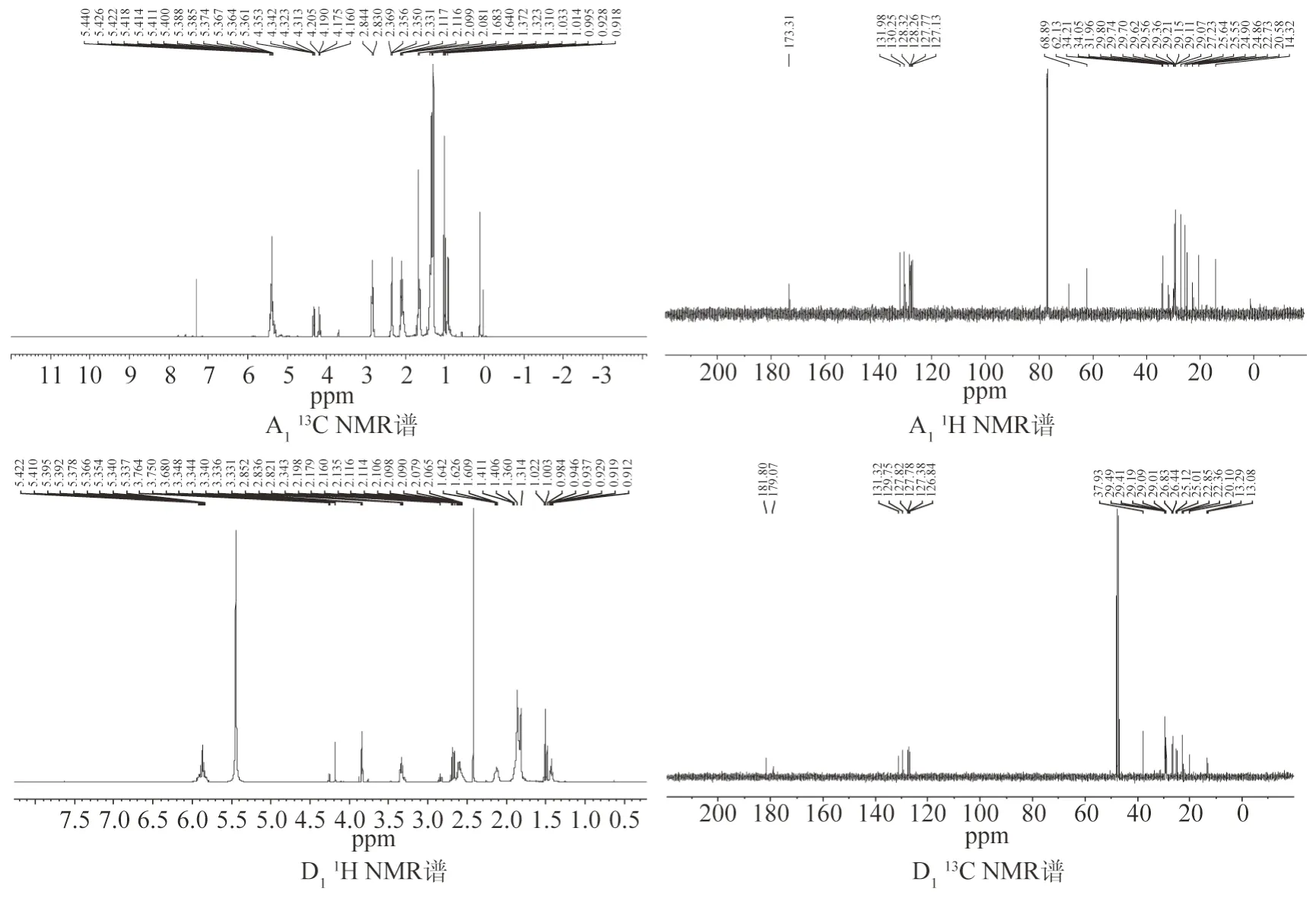
圖3 A1,D1的1H和13C NMR譜Fig.3 1H and 13C NMR spectra of A1 and D1
結合質譜與核磁結果,鑒定五種物質結構式如下所示(圖4)。推測其中物質A1是葉綠素b在酸性條件下,中心鎂離子丟失,被氫離子取代,且第Ⅱ吡咯環外的甲基氧化成羰基;物質B1是脫鎂葉綠酸a副環上的羧基在酸性條件下與氫離子反應,生成羧酸;物質D1推測的結構與脫鎂葉綠酸a一致,證明該組分物質為脫鎂葉綠酸a。物質E1是葉綠素a的α位開環,β和δ為加上兩個氧,與Vergeiner等[19]研究中的葉綠素熒光與非熒光降解產物的共同前體(epi-pfcc)結構類似,唯一區別在于物質E1的葉綠醇并未斷裂,說明該物質結構是生成的降解產物共同前體反應前一步的結構,與降解過程相符合;物質F1是葉綠素a在用乙酸乙酯、甲醇和乙醇提取過程中,α位被氧化開環,葉綠醇被乙酸取代,在酸性條件下鎂離子丟失,第I吡咯環外的烯基被加上了兩個氧,其結構與文獻[16,20]中葉綠素非熒光降解產物(Hv-NCC-1)的結構極其相似,區別在于物質F1第Ⅱ吡咯環外的甲基并未降解變化為羰基,且第Ⅳ吡咯環側鏈上的丙酸與乙酸乙酯發生反應。根據此推測結果與文獻進行參考,綜合葉綠素穩定性因素考慮,進行葉綠素提取和分離純化時,應在密封、pH中性等條件下進行,保障提取和純化時葉綠素結構的穩定性,因葉綠素降解同時需要酶的參與,也可通過改變條件影響酶活,從而達到保護其結構穩定的目的[21-22]。
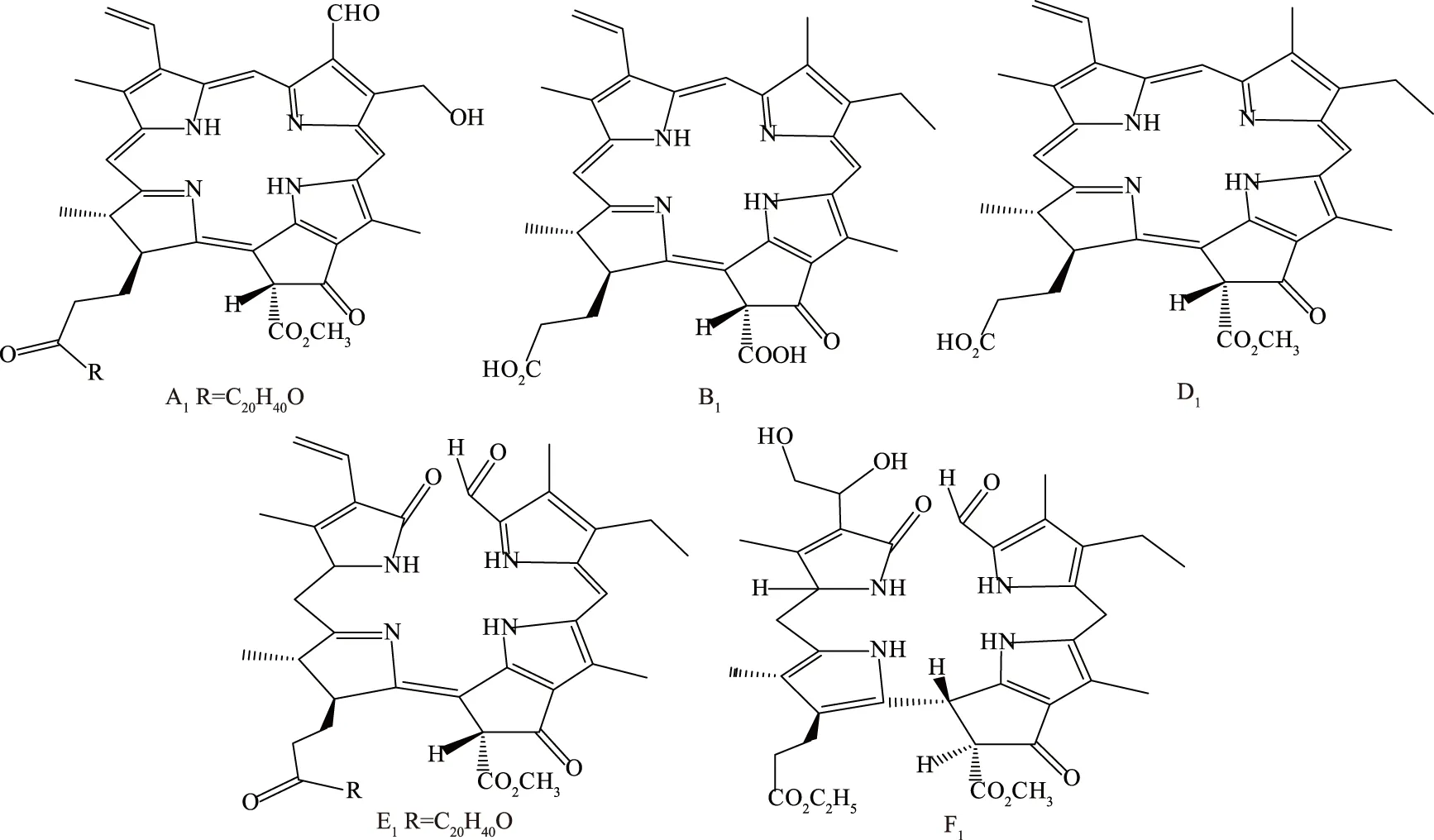
圖4 五種物質的結構式Fig.4 Structural formulas of five kinds of substances
2.4 純化物的抗氧化活性分析
2.4.1 純化物對DPPH·清除作用 由圖5可知,獼猴桃中各提取物對DPPH·的清除作用隨純化物質量濃度增大而增加,在所測的各提取物中,物質C1、D1、E1、F1對DPPH·清除效果較好,物質B1的清除效果稍差,在質量濃度為0.12 mg/mL時清除率為77.47%,活性最差的是物質A1,當質量濃度為0.12 mg/mL時清除率為13.97%。
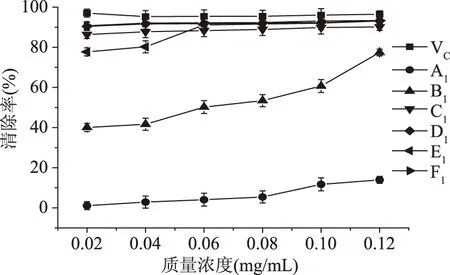
圖5 獼猴桃提取物對DPPH·清除率Fig.5 Scavenging rates of different solvent extracts from Kiwifruit against DPPH· free radicals
2.4.2 純化物對DPPH·清除活性IC50值的影響 由表1可見,提取物對DPPH·清除活性IC50值的順序為:VC<物質E1<物質C1<物質F1<物質D1<物質B1<物質A1,IC50越小說明對 DPPH·的清除效果越好,在各種提取物中C1與E1的活性最強,略低于VC,提取活性最低的是A1,為純石油醚分離產物。

表1 獼猴桃提取物對DPPH·清除活性的IC50值Table 1 IC50 values of different solvent extracts from kiwifruit for scavenging DPPH· free radicals
2.5 提取物對Fe3+還原力的測定
由圖6可知,獼猴桃中提取物對Fe3+有較強的還原能力,物質C1、E1、F1的還原能力與VC近似,物質D1的還原能力稍弱,物質A1和B1的還原能力明顯弱于VC,與DPPH·清除實驗結果基本一致。
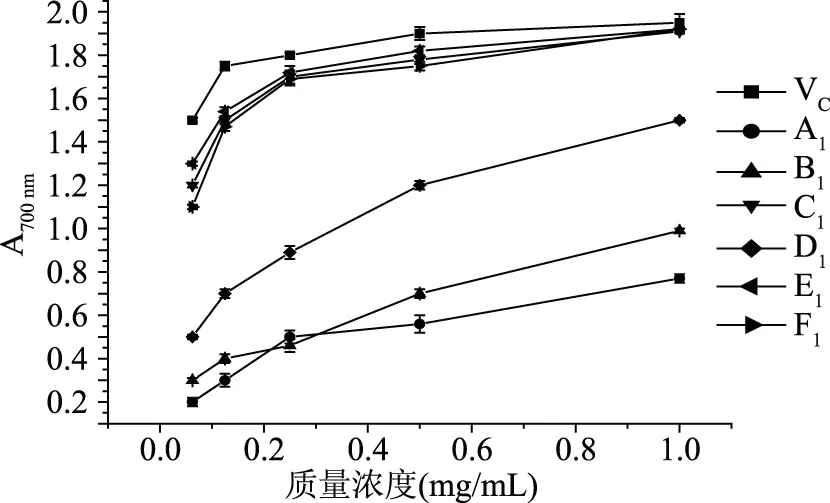
圖6 獼猴桃提取物對Fe3+還原力曲線Fig.6 Ferric reducing power curves of different solvent extracts from Kiwifruit
3 結論
利用溶劑提取法和柱層析等手段分離純化得到包括葉綠素a在內的6種高純度物質,通過質譜得到了它們[M+H]+的準確分子質量,結合核磁共振數據推測了5種葉綠素相關物質的結構。其中C1為葉綠素a,得到物質A1的[M+H]+的分子質量為927.6967,分子式為C55H84O7N4;物質B1的[M+H]+的分子質量為588.4304,分子式C34H33O5N4;物質D1的[M+H]+的分子質量為601.1116,分子式為C35H34O5N4,推測結構可知為脫鎂葉綠酸a;物質E1的[M+H]+的分子質量為927.6985,分子式C55H84O7N4;物質F1的[M+H]+的分子質量為701.5039,分子式C37H38O9N4。同時通過DPPH法和Fe3+還原力法對其單品進行了抗氧化活性研究,結果顯示E1與F1的活性最強,在質量濃度為0.12 mg/mL時,DPPH·清除率分別為93.14%和93.39%,略低于VC的96.49%;在質量濃度為1.0 mg/mL時,物質E1與F1的吸光值均為1.92,僅次于VC的1.95,說明抗氧化性強,與清除實驗結果相吻合。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
