改良比色法檢測食品中的亞硝酸鹽
2019-02-20 02:58:32苗攀登劉鐘棟
食品工業科技 2019年23期
關鍵詞:檢測
苗攀登,劉鐘棟
(河南工業大學糧油食品學院,河南鄭州 450001)
亞硝酸鹽是一種很常見的食品添加劑和天然衍生物[1],在香腸、果醬、腌制品、肉脯等食品中都有不同劑量的添加,主要是起著色和防腐的作用。亞硝酸鹽是劇毒物質,其成人中毒劑量為0.2~0.5 g,致死劑量為3 g。在胃酸等環境下亞硝酸鹽與食物中的仲胺、叔胺和酰胺等反應生成強致癌物N-亞硝胺,它有極強的致癌作用。亞硝酸鹽還能使血液中正常攜氧的低鐵血紅蛋白氧化成高鐵血紅蛋白,因而失去攜氧能力而引起組織缺氧。此外,亞硝胺還能夠透過胎盤進入胎兒體內,對胎兒有致畸作用。6個月以內的嬰兒對亞硝酸鹽特別敏感,臨床上患“高鐵血紅蛋白癥”的嬰兒即是食用亞硝酸鹽濃度高的食品引起的,癥狀為缺氧,出現紫紺,甚至死亡[2-4]。
食品在生產、銷售等過程中,均存在著各種各樣的安全隱患,其中亞硝酸鹽超標是一個非常嚴重的問題。亞硝酸鹽的危害無需贅述,所以詳細記錄低值食品原料中的亞硝酸鹽含量的變化,是控制亞硝酸鹽含量的必要手段。現階段的檢測手段有光度法、化學發光法、電化學法、色譜法、毛細管電泳法。最常見的光度法即是比色法,其原理是:在酸性環境下,對氨基苯磺酸首先與亞硝酸根反應生成重氮化合物,然后再與N-(1-萘基)-乙二胺發生偶合反應,形成紫紅色偶氮化合物,在540 nm波長處具有最大吸收且與亞硝酸鹽含量呈正相關,檢測限為0.06 μg/mL。此方法已經非常成熟、重復性好,準確度高,但步驟繁雜,易受其它雜質干擾,試劑毒性較大,也鑒于其檢測限的限制,近些年逐步被離子色譜法取代[5-9]。化學發光法的原理是根據亞硝酸鹽濃度與分子發光強度呈線性關系,通過檢測分子發光強度來得到亞硝酸鹽的濃度。此方法具有操作方便靈敏度高、試劑消耗少、儀器設備簡單等優點,但也有實驗儀器昂貴、操作人員專業要求高、工作環境要求高等缺陷[10-11]。電化學方法中常見的極譜法,是利用電解過程中所得到的極化電極的電流電位曲線來確定溶液中物質濃度的電化學分析方法,具有設備簡單、分析速度快、準確度高、靈敏度高的優點,但是有設備昂貴、不易保養以及重現性差等缺陷[12-13]。離子色譜法是利用陰陽離子對載體(樹脂)的吸附能力不同達到分離的目的[14-16]。總的來說,色譜法具有線性范圍寬、抗干擾能力強、操作簡便特點,但有設備儀器昂貴、操作人員要求高、耗時等缺點;此外,亞硝酸鹽的檢測方法還有毛細管電泳法、催化光度法、熒光光度法、流動注射分光光度法、電化學伏安法、氣相色譜法以及高效液相色譜法等。光度法使用方便但是不夠靈敏,容易受干擾,其他方法雖有達到較低檢測限,但是設備昂貴,不易保養,操作繁雜,不宜大批檢測樣品,又或者耗時長,時效性不高。因此改進光度法,使其克服固有的缺點是一個建立新方法的思路。
在本研究中,將對氨基苯硫酚(ATP)和萘基乙二胺(NED)分別修飾在納米金顆粒上之后,在酸性條件下,再加入亞硝酸鹽,體系將會發生重氮化耦合反應,使金納米顆粒發生聚集,從而導致體系光學性質(顏色)發生變化。通過探究顏色變化與亞硝酸鹽濃度的關系來建立亞硝酸鹽檢測方法,計算檢測限。
1 材料與方法
1.1 材料與儀器
面包 包頭市味多美食品有限公司;火腿 河南雙匯投資發展股份有限公司;海帶絲 山東煙臺萬歷海藻食品有限公司;袋裝牛奶 內蒙古伊利實業集團股份有限公司;瓶裝水 康師傅控股有限公司;實驗所用食品材料 為市場隨機購買所得;水合氯金酸(HAuCl4) Sigma Aldrich公司;檸檬酸鈉、對氨基苯磺酸、對氨基苯硫酚ATP、鹽酸萘乙二胺NED、巰基乙酸 北京化學試劑公司;亞硝酸鈉標準溶液(1000 μg/mL) 成都博瑞特化學技術有限公司,可直接使用無需校準;濃鹽酸、氫氧化鈉 天津市大茂化學試劑廠;無水乙醇、亞鐵氰化鉀、乙酸鋅 麥克林試劑有限公司;去離子水 天津卓精虹科儀器設備技術有限公司;其他化學試劑 均為分析純。
U-2900型紫外分光光度計 Hitachi;ZS90型粒度電位儀 Malvern;瓊脂糖電泳設備 天津市華茂電泳設備有限公司;磁力攪拌器及其配套電熱套 鞏義市予華儀器設備有限公司;小型振蕩器 上海亞榮生化儀器廠;離心機 上海盧湘儀;JR05-300型食品粉碎機 浙江蘇泊爾股份有限公司;KQ-100DE型超聲波儀(輸出功率:400 W)昆山市超聲儀器有限公司以及其他常規實驗室器皿。
1.2 實驗方法
1.2.1 樣品前處理方法 取面包、火腿、海帶絲各0.25 g分別放入食品粉碎機再加入10 mL水,粉碎25 min,并轉移到25 mL容量瓶中,三次用少量水(2 mL左右)清洗粉碎機杯壁倒入容量瓶中,再定容到25 mL;瓶裝水和袋裝牛奶直接轉移到容量瓶并定容到25 mL[17-19]。所有樣品定容后,在超聲清洗機中(400 W)浸泡30 min,然后2000 r/min離心15 min,取上清液5 mL(其中瓶裝水樣品無分層),加入200 μL亞鐵氰化鉀溶液(106 g/L),再加入0.5 mL乙酸鋅溶液(220 g/L),手動攪拌5~10 min,過濾得到樣品液[20];用體系檢測樣品液亞硝酸鹽濃度c(μg/mL),即樣品的亞硝酸鹽含量:C=c×25/0.25(μg/g),C=100c。
1.2.2 傳統比色法檢測亞硝酸鹽
1.2.2.1 傳統比色法的標準曲線的繪制 參考國標GB 5009.33-2016,1000 μg/mL的亞硝酸鈉標準液用去離子水分別稀釋到0.05、0.1、0.5、1.0、3.0、5.0、10.0、15.0、20.0 μg/mL,再分別取4.7 mL稀釋液和去離子水(空白),分別放在試管中,加入0.2 mL 4 g/L對氨基苯磺酸溶液,混勻,靜置3~5 min后各加入1 mL 2 g/L鹽酸萘乙二胺溶液,混勻靜置10 min。用1 cm比色杯,以空白管調節零點,于波長538 nm處測吸光度,繪制標準曲線[21]。

1.2.2.3 傳統比色法的加標回收試驗 以正常銷售的鮮牛奶和瓶裝水為空白,分別取鮮牛奶和瓶裝水2.5 g。向兩種樣品中分別加入1.00、5.00、10.00、20.00 μg/mL的亞硝酸鹽標準品,用去離子水定容25 mL,在輸出功率400 W的超聲儀中浸泡30 min;離心取上清液5 mL,加入200 μL亞鐵氰化鉀溶液(106 g/L),再加入0.5 mL乙酸鋅溶液(220 g/L),攪拌5~10 min,過濾得到樣品液,然后用傳統比色法檢測樣品液的亞硝酸鹽濃度c,即亞硝酸鹽的添加量m=c×25,計算出回收率并重復10次計算相對標準偏差。
1.2.3 改良比色法檢測亞硝酸鹽
1.2.3.1 實驗原理 利用金納米顆粒優秀的光學性質,將對氨基苯硫酚通過S-Au鍵修飾到水相金納米顆粒記為ATP-GNPs;將萘基乙二胺(NED)通過1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(EDC)催化,連接到雙親性聚合物包裹的油相金納米顆粒(AP-GNPs)上得到萘基乙二胺修飾的金納米顆粒記為NA-GNPs,在酸性條件下,加入NO2-后,發生重氮化耦合反應,兩種修飾過的金納米顆粒被連在一起,發生聚合,金納米顆粒的顏色發生明顯變化,這種變化與NO2-的濃度呈良好線性關系。總之,改進比色法用金納米顆粒的靈敏的光學變化代替了耦合反應生成的染料分子的顏色變化,使體系的檢測線更低,靈敏度更高,如圖1。

圖1 改良比色法基本原理示意圖Fig.1 Schematic diagram of the the improved colorimetric basic principle
1.2.3.2 ATP-GNP的制備及表征 取17 mg水合氯金酸溶于50 mL的去離子水中,沸騰回流10 min,溶液呈淡黃色;78 mg檸檬酸鈉溶于5 mL去離子水中,預熱到60~70 ℃,快速加入到回流后的水合氯金酸的溶液中,繼續加熱回流20 min,溶液顏色由黃色變無色隨后顏色逐漸加深,最后呈透亮的深紅色,冷卻至室溫,調節pH到7,用0.45 μm的濾膜過濾兩次,室溫備用,這時的金納米顆粒GNPs的粒徑大約為10 nm[22]。由郎伯比爾定律(式1)計算得到的GNPs的吸光度為A=1.924,可計算得知GNPs的濃度c=1.40×10-8mol/L。
郎伯比爾定律A=ε×C×l
式(1)
式中:A是吸光度值,ε是吸收常數,l是光徑長度(cm,所使用的儀器光徑長度規格為1 cm),ε10 nm=1.37×108cm-1mol-1L。
將制備好的金納米顆粒稀釋10倍,將pH調整到弱酸性(pH=3~4),用10 mmol/L的ATP(溶解于無水乙醇)與稀釋后的納米金顆粒按體積1∶8的比例混合均勻,在搖床上勻速振蕩6 h,反應后,用5000 r/min離心30 min,去上清,再用去離子水溶解,反復3~5次,即得到ATP-GNPs。ATP-GNPs的粒徑約為10 nm,由郎伯比爾定律[23-24],計算出ATP-GNPs濃度約為10-9mol/L。
制備ATP-GNPs之后,在室溫下利用粒徑電位儀、全波譜紫外分光光度計表征GNPs和ATP-GNPs的粒徑分布、電位變化以及吸收圖譜的變化,其中樣品皿的規格為:1×1 cm的石英皿。
1.2.3.3 NA-GNPs的制備及表征 有機相金納米顆粒是在有機相中用硼氫化鈉快速還原氯金酸得到的,再利用兩親性的聚合物的包裹使其“溶解”到水相中,再通過反應將萘基乙二胺連接到兩親性聚合物的羧基上,最終得到萘基乙二胺修飾的金納米顆粒(NA-GNPs),詳細反應步驟見a~d。
a:有機相合成金納米顆粒GNPs:取2.17 g四辛基溴化銨溶于80 mL甲苯中,在輸出功率400 W的超聲儀中浸泡2~3 min使四辛基溴化銨完全溶解,溶液澄清透明無雜質[25]。取300 mg氯金酸溶于25 mL純水中,溶液為澄清的黃色。上述兩種溶液混合,輕搖直到水相中的黃色完全消失,有機相變成紅色。用分液漏斗將水油分開,有機相轉移到燒瓶中。334 mg硼氫化鈉溶于25 mL純水。硼氫化鈉溶液在1 min之內加入之前含有金的甲苯溶液中,邊攬拌邊滴加。然后金溶液在室溫下攪拌l h。攪拌l h之后,再利用分液漏斗溶液將水油分開,有機相先用10 mmol/L鹽酸洗3遍,再用10 mmol/L氫氧化鈉洗3遍。將溶液轉到燒瓶中攪拌12 h后加入10 mL十二硫醇,65 ℃水浴攪拌3 h。所得溶液2000 r/min離心5 min,收集上清,去除雜質。再按1∶1加入甲醇2000 r/min離心5 min。去除含有四辛基溴化銨和十二硫醇的上清液,收集GNPs沉淀溶于氯仿中。合成的GNPs粒徑約5 nm。
b:制備兩親性聚合物:2.7 g十二胺和3.084 g聚(異丁烯-alt-馬來酸酐)溶于100 mL四氫呋喃,55~60 ℃水浴攪拌l h。此過程四氫呋喃會揮發,溶液大概濃縮到30~40 mL。停止加熱,剩余溶液在室溫下攪拌12 h。利用真空旋轉蒸發儀使四氫呋喃完全揮發,加入40 mL氯仿劇烈攪拌,使瓶底附著的物質完全溶解,最終所得兩親性聚合物AP。
c:AP包被GNPs:取1 mL GNPs(溶于氯仿)于圓底燒瓶,再加入0.2 mL AP,搖晃均勻,加入5 mL的氯仿降低體系濃度。緩慢抽真空使氯仿揮發直到混合物完全干燥。加入適量SB12(0.05 mol/L硼酸緩沖液,pH=12)劇烈攪拌,使粘附在瓶壁上的物質完全溶解。用60000 r/min離心30 min進行純化,去上清用去離子水復溶,重復3~5次,就得到兩親性聚合物包被的金納米顆粒,記為AP-GNPs,如圖2。
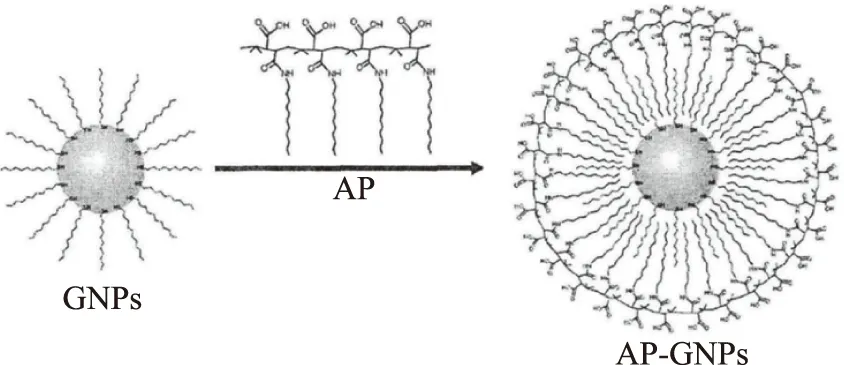
圖2 AP包被GNPs的示意圖Fig.2 Scheme of AP coating on GNPs
d:萘基乙二胺修飾AP-GNPs得到NA-GNPs:10 mL 20 mmol/L鹽酸萘乙二胺溶液(溶于硼酸緩沖液,pH調到9)與1 mL AP-GNPs混勻,再加入1 mL 0.5 mol/L的EDC,混勻靜置3 h。用60000 r/min離心30 min進行純化,去上清用去離子水復溶,重復3~5次,就得到NA-GNPs,其粒徑約為10 nm。由郎伯比爾定律,計算出NA-GNPs濃度約為210-7mol/L。將NA-GNPs稀釋20倍后,與ATP-GNPs等體積混合,將體系pH調整到5,作為反應液存放于-4 ℃備用。將亞硝酸鈉標準溶液分別稀釋到0.005、0.01、0.05、0.1、0.5、1、2、3、4、5 μg/mL。分別取濃度梯度的標準液和去離子水(空白)0.5 mL,再加入0.5 mL反應液,混勻靜置15~20 min,測體系的吸光度,繪制標準曲線。
e:表征:在室溫下利用粒徑電位儀、全波譜紫外分光光度計表征油相GNPs、AP-GNPs和NA-GNP的粒徑分布、電位變化以及吸收圖譜的變化,其中樣品皿的規格為:1×1 cm的石英皿。
1.2.4 存放時間和其他離子對檢測體系的影響 將反應液存放1、2、3、4、5周,分別測紫外吸光度,觀察金納米顆粒520 nm處的特征吸收峰,證明反應液的穩定性。
1.2.5 改良比色法的加標回收試驗 以正常市售的鮮牛奶和瓶裝水為空白,分別取鮮牛奶和瓶裝水2.5 g。向兩種樣品中分別加入5、10、20 μg的亞硝酸鹽標準品,定容25 mL,攪拌超聲浸泡30 min;以2000 r/min離心15 min取上清液5 mL,加入200 μL亞鐵氰化鉀溶液(106 g/L),再加入0.5 mL乙酸鋅溶液(220 g/L),攪拌5~10 min,過濾得到樣品液,然后用改良比色法檢測樣品液的亞硝酸鹽濃度c,即亞硝酸鹽的添加量m=c×25,計算出回收率并重復10次計算相對標準偏差[26-27]。
1.2.6 兩種比色法的實際應用 將面包、火腿、海帶絲打開包裝,放置在常溫(25 ℃)下0、5、10、15、20、25、30、35、40 h,分別取0.25 g各個時段的面包、火腿、海帶絲,按照1.2.1制備樣品液,C樣品=100c樣品液(μg/g)。用傳統比色法和改良比色法檢測樣品液,對比兩種比色法的差異。
2 結果與分析
2.1 傳統比色法檢測亞硝酸鹽
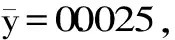

圖3 傳統比色法的標準曲線和各樣品的顏色變化Fig.3 Standard curve of traditional colorimetric method and the color changes of the samples

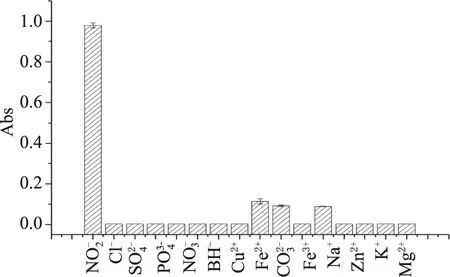
圖4 不同離子對體系的影響Fig.4 Effect of different ions on the reaction solution
2.1.3 傳統比色法的加標回收試驗 按照1.2.2.3的方法,傳統比色法的加標回收試驗結果如表1。由表1可見,在加入1.00 μg/mL時,傳統比色法無法檢測出準確的亞硝酸鹽含量,顯然加標樣品的亞硝酸鹽含量已經低于其檢測限,因此傳統比色法在應用方面有檢測限的局限。在5.00、10.00、20.00 μg/mL的加標樣品的回收率為97.88%~100.24%,相對標準偏差(RSD)為3.53%~7.22%。
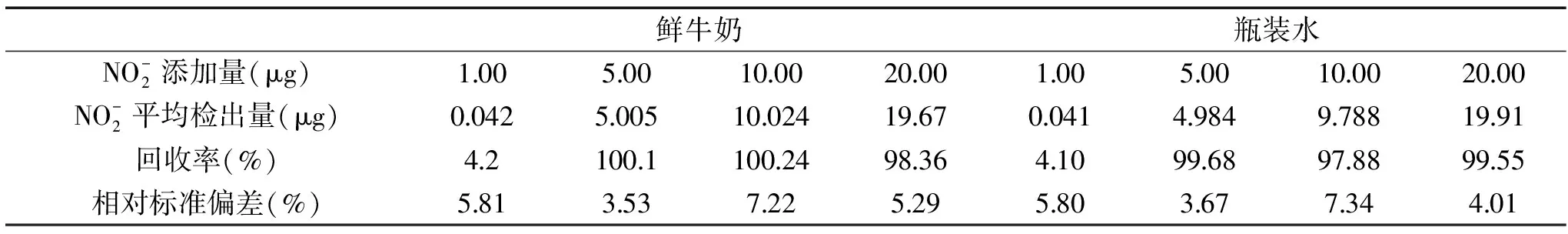
表1 傳統比色法的回收率以及相對標準偏差Table 1 Recovery rate and relative deviation(RSD)of the traditional colorimetric method(n=10)
2.2 ATP-GNPs的表征
通過1.2.3.2的方法制備出水相金納米顆粒(GNPs),對氨基苯硫酚(ATP)修飾到金納米顆粒之后,由于金納米顆粒表面配體的變化,紫外吸收峰有少許偏移,純化過程有少量損失,所以吸收峰強度下降,如圖5;表面電位也有變化,如圖6;ATP-GNPs的粒徑約是10 nm,如圖7。

圖5 ATP修飾GNPs前后紫外吸收圖譜的變化Fig.5 Changes in UV absorption spectra before and after ATP-modified GNPs
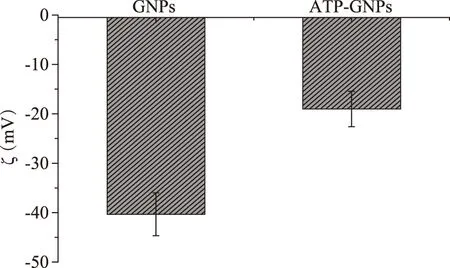
圖6 ATP修飾GNPs前后表面電位的變化Fig.6 Changes in surface potential before and after ATP-modified GNPs

圖7 ATP-GNPs的粒徑表征Fig.7 Particle size characterization of ATP-GNPs
2.3 油相GNPs、AP-GNPs、NA-GNPs的表征
用1.2.3.3方法步驟制作出油相GNPs的粒徑約為5 nm,用AP包被后的AP-GNPs的粒徑約為10 nm,而且紫外吸收吸收峰發生偏移;由于疏水性,油相GNPs被AP包被,親水的羧基暴露在外側,在EDC的催化下與萘基乙二胺(NED)的氨基連接在一起。純化后把多余的NED去除,就得到了NA-GNPs,由于NED的分子較小,因此NA-GNPs的紫外吸收峰相對于AP-GNPs幾乎沒有變化,粒徑的變化也不大,只有純化過程中有損失,峰強度降低。油相GNPs、AP-GNPs、NA-GNPs的紫外吸收圖譜、粒徑,如圖8、圖9。
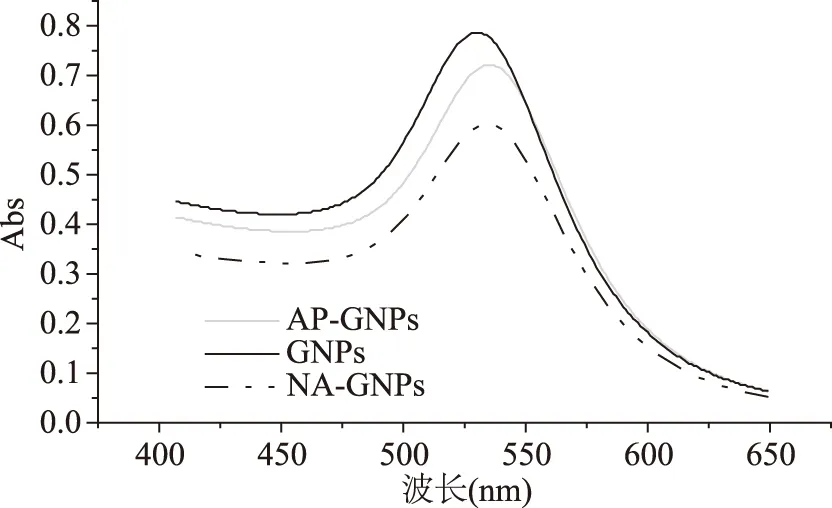
圖8 油相GNPs、AP-GNPs、NA-GNPs的紫外吸收圖譜Fig.8 Ultraviolet absorption spectra of oil phase GNPs,AP-GNPs and NA-GNPs
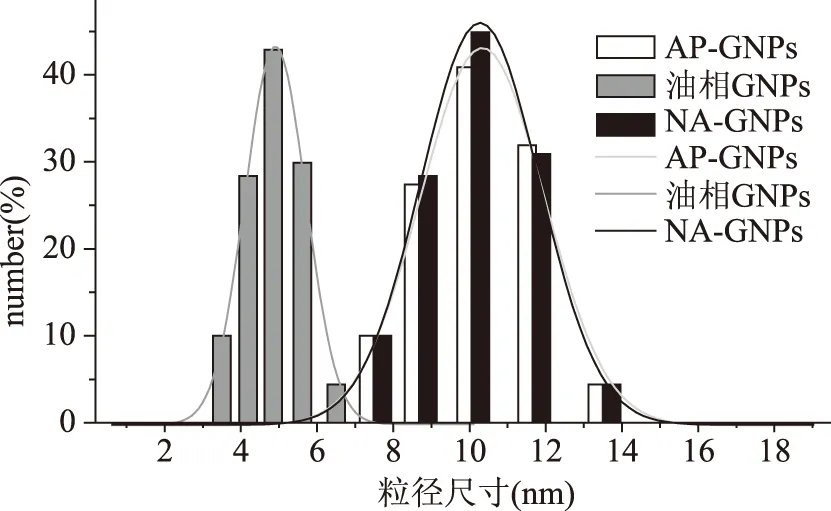
圖9 油相GNPs、AP-GNPs、NA-GNPs的粒徑表征Fig.9 Size characterization of oil phase GNPs,AP-GNPs and NA-GNPs
2.4 改良比色法的結果分析


圖10 加入濃度梯度溶液后體系的紫外吸收圖譜Fig.10 UV absorption spectrum of the system by adding a concentration gradient solution

圖11 濃度梯度的的標準曲線Fig.11 Standard curve of concentration gradient
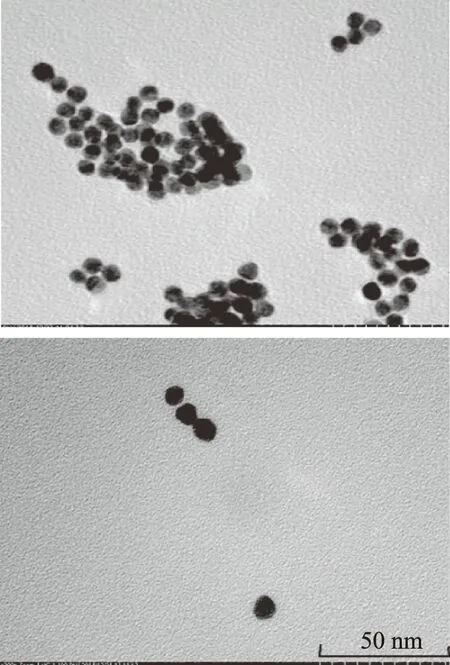
圖12 同等條件下,加入溶液前(右)和后(左),金納米顆粒的聚集情況Fig.12 Aggregation of gold nanoparticles before(right)and after(left)addition of solution under equivalent conditions
利用空白樣品液10次測量吸光度得到10個A580 nm/A520 nm的值,其標準偏差為0.11%。由公式dl=3σ/k,其中dl是檢測限,σ是指空白樣品的標準偏差,k是指標準曲線的斜率。通過計算體系的檢測限dl=5.12 ng/mL。
2.5 存放時間和其他離子對檢測體系的影響
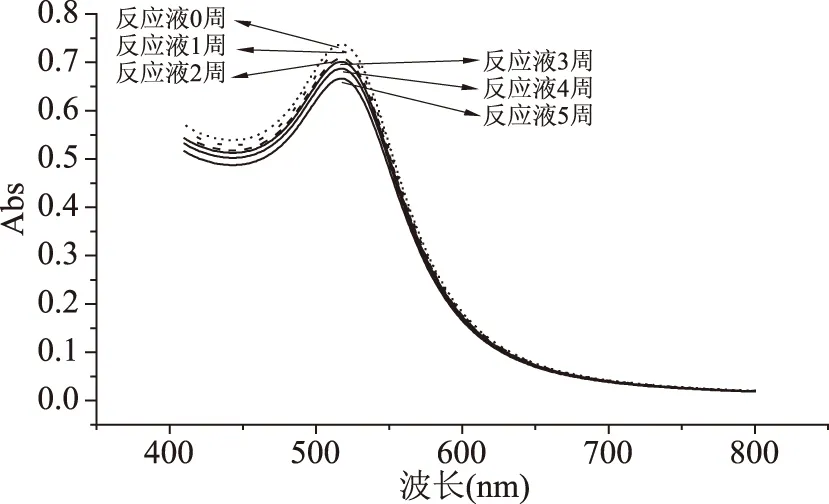
圖13 不同存放時間,反應液的紫外吸收圖譜Fig.13 UV absorption spectrum of the reaction solution at different storage time

圖14 不同離子對反應液的影響Fig.14 Effect of different ions on the reaction solution
2.6 改良比色法的加標回收試驗
按照1.2.5的方法,改良比色法的加標回收實驗結果如表2。
由表2可見,樣品回收率為97.00%~100.28%,相對標準偏差(RSD)為0.77%~3.88%,表明本實驗方法具有可靠地準確度和精密度。
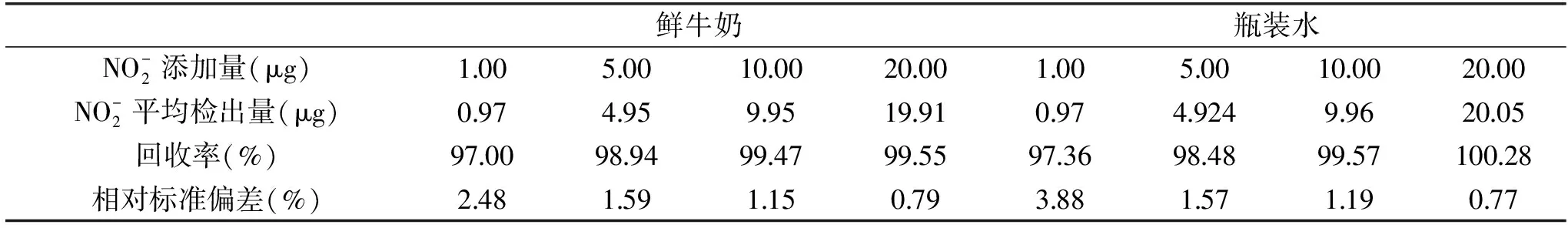
表2 改良比色法的回收率以及相對標準偏差Table 2 Spike recovery rate and relative deviation(RSD)of the improved colorimetric method(n=10)
2.7 改良比色法與傳統比色法的比較
2.7.1 檢測范圍和檢出限的比較 兩種方法的檢測范圍差異很大,表明了改良比色法檢測極微量的亞硝酸鹽方面的優勢,但是傳統比色法檢測范圍更廣。改良比色法的檢測限低至5.12 ng/mL,比傳統比色法靈敏度更高。在2.6中也能得到證實,加標1.00 μg的樣品處理液的濃度為40 ng/mL,傳統比色法無檢測信號,然而改良比色法能夠正常的檢測其濃度,因此顯示出改良比色法的優勢。
2.7.2 回收率的比較 改良比色法和傳統比色法各自的檢測范圍內的回收率相差無幾,約為97%~100%,說明兩種方法在各自的檢測范圍內都有良好的準確性和可靠性;傳統比色法的RSD為3.53%~7.22%,明顯比改良比色法大,說明改良比色法檢測結果的離散度程度較小,即檢測穩定性比傳統比色法優秀。
2.7.3 響應時間的比較 傳統比色法是先用對氨基苯磺酸與亞硝酸根發生重氮化反應(3~5 min),再與萘基乙二胺鹽酸鹽發生耦合顯色(15 min),整個過程需要15~20 min,而改良比色法的對氨基苯硫酚和萘基乙二胺連接到金納米顆粒后,使用時只加入亞硝酸鈉溶液即可,這一過程約15 min,所以改良比色法操作簡單,節約反應時間,提高效率。

表3 改良比色法與傳統比色法的比較Table 3 Comparison between improved colorimetry and traditional colorimetry
2.7.4 穩定性的比較 改良比色法的兩種金納米顆粒混合后可以穩定存放5周以上,Cu2+、Fe2+、Fe3+對新舊兩種檢測體系都有少許影響,可能是因為較高濃度的干擾離子本身的顏色對檢測體系的吸光度有一些影響;總之改良比色法具有良好的穩定性和抗離子干擾的能力。
2.7.5 相對標準偏差RSD的比較 傳統比色法的RSD為3.53~7.22,明顯比改良比色法大,說明改良比色法檢測結果的離散度程度較小,即檢測穩定性比傳統比色法優良。
總的來說,與傳統比色法相比,改良比色法具有檢測限低、靈敏度高、操作簡單、檢測效率高、穩定性好等優勢,但是檢測范圍窄是改良比色法的缺點。改良比色法適用于極微量的亞硝酸鹽的檢測,如飲用水、牛奶、嬰兒食品等亞硝酸鹽含量允許添加量極低的食品的檢測,或者研究食品中亞硝酸鹽的產生過程。
2.8 兩種比色法的實際應用
查閱中國的食品標準,面包的允許殘留量為2 g/mg,火腿的允許殘留量為70 μg/mg,海帶絲的允許殘留量為20 μg/mg。如圖15,面包的亞硝酸鹽含量較低,傳統比色法檢測時,30 h之前無法響應,只有亞硝酸鹽增加到傳統比色法的檢測限后,才能有響應。然而改良比色法檢測樣品時,能夠很好地反應亞硝酸鹽的增長過程;火腿的亞硝酸鹽含量較高,如圖16,20 h后,樣品的亞硝酸鹽含量就達到了新方法的檢測極限,于是體系處于飽和狀態,無法準確記錄后面的樣品。火腿中亞硝酸鹽含量較高,而且暴露15 h后,亞硝酸鹽增速加大,25 h左右就超過了允許殘留量,如圖17。海帶絲在工廠腌制過程中,一般都存放超過20 d,產品中有一定的乳酸菌消耗了亞硝酸鹽,因此樣品的亞硝酸鹽含量增速較慢,但是也不宜存放超過30 h。總之,改良比色法優化了檢測限,但是檢測范圍較窄(0~1 μg/mL),因此改良比色法適用于極低含量亞硝酸鹽的測定,如奶制品、面制品等;另一方面,改良比色法可以用于環境污染監測,如水庫、江河水的亞硝酸鹽含量的檢測等。

圖15 面包中的亞硝酸鹽的含量變化Fig.15 Changes in nitrite content in bread

圖16 火腿中的亞硝酸鹽的含量變化Fig.16 Changes in nitrite content in ham
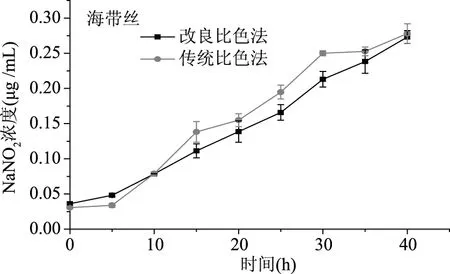
圖17 海帶絲中的亞硝酸鹽的含量變化Fig.17 Changes in nitrite content in seaweed strips
3 結論
在本研究中,利用功能化金納米顆粒對傳統比色法進行了改良。相比傳統比色法,改良比色法的檢測限達到了5.12 ng/mL,遠遠低于傳統比色法(76.43 ng/mL)。改良比色法的操作步驟簡便、響應時間短,大大提高了工作效率。雖然Cu2+、Fe2+、Fe3+對傳統比色法和改良比色法都有影響,但是改良比色法在回收率、RSD方面有很大優勢,說明此方法的穩定性更高,更能適應更復雜的工作環境。改良比色法的檢測范圍較窄,更加適用于微量亞硝酸鹽含量的檢測,是傳統比色法的有效補充和改進。在應用中發現,食品打開包裝后,應盡快用完,否則有亞硝酸鹽超標的危險。改良比色法具有高靈敏性的特點,有利于研究亞硝酸鹽產生的過程和原因,以及預防亞硝酸鹽的產生。
權威·高效·核心·領先·精湛·實用
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
